

DNAJC6 Mutations Disrupt Dopamine Homeostasis in Juvenile Parkinsonism-Dystonia
- PMID: 32472658
- PMCID: PMC8425408
- DOI: 10.1002/mds.28063
DNAJC6 Mutations Disrupt Dopamine Homeostasis in Juvenile Parkinsonism-Dystonia
Abstract
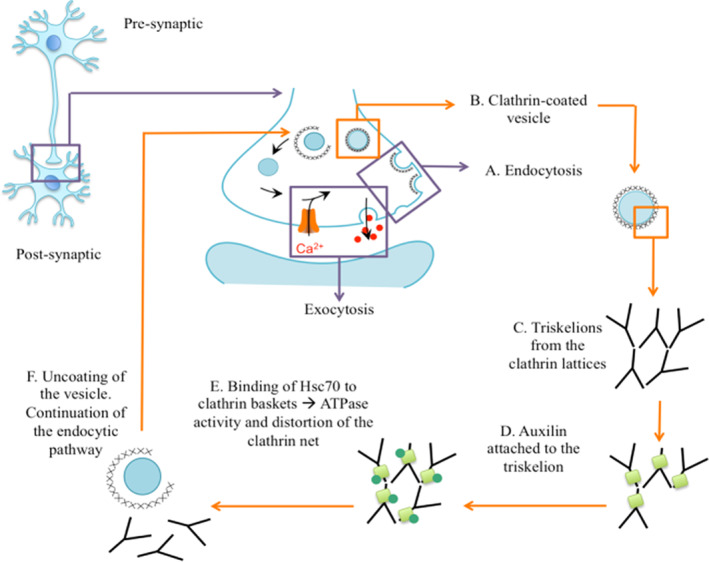
Background: Juvenile forms of parkinsonism are rare conditions with onset of bradykinesia, tremor and rigidity before the age of 21 years. These atypical presentations commonly have a genetic aetiology, highlighting important insights into underlying pathophysiology. Genetic defects may affect key proteins of the endocytic pathway and clathrin-mediated endocytosis (CME), as in DNAJC6-related juvenile parkinsonism.
Objective: To report on a new patient cohort with juvenile-onset DNAJC6 parkinsonism-dystonia and determine the functional consequences on auxilin and dopamine homeostasis.
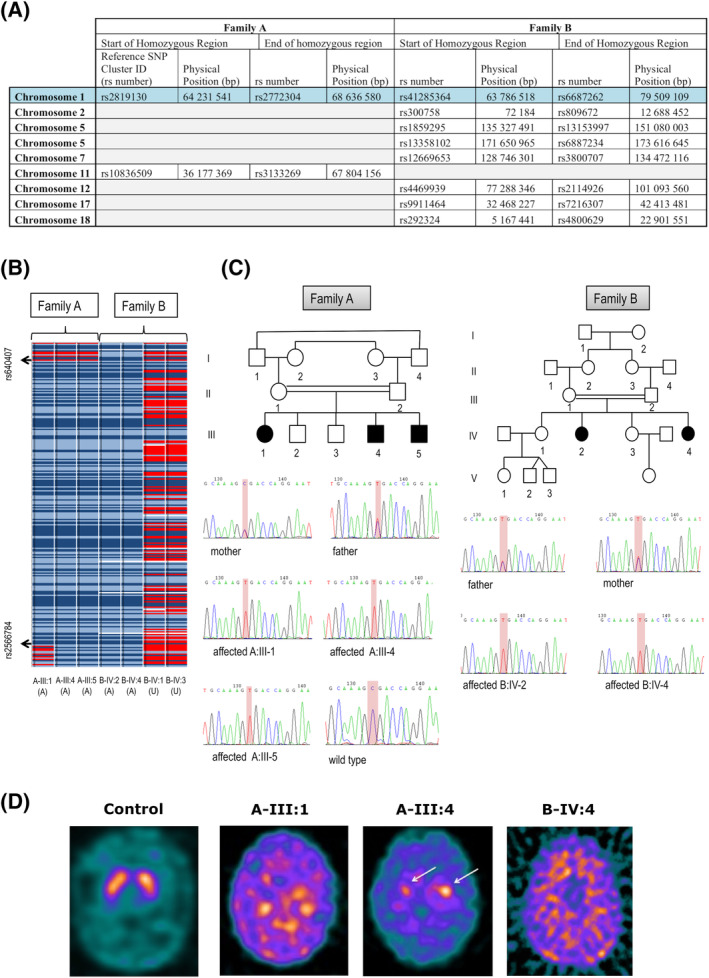
Methods: Twenty-five children with juvenile parkinsonism were identified from a research cohort of patients with undiagnosed pediatric movement disorders. Molecular genetic investigations included autozygosity mapping studies and whole-exome sequencing. Patient fibroblasts and CSF were analyzed for auxilin, cyclin G-associated kinase and synaptic proteins.
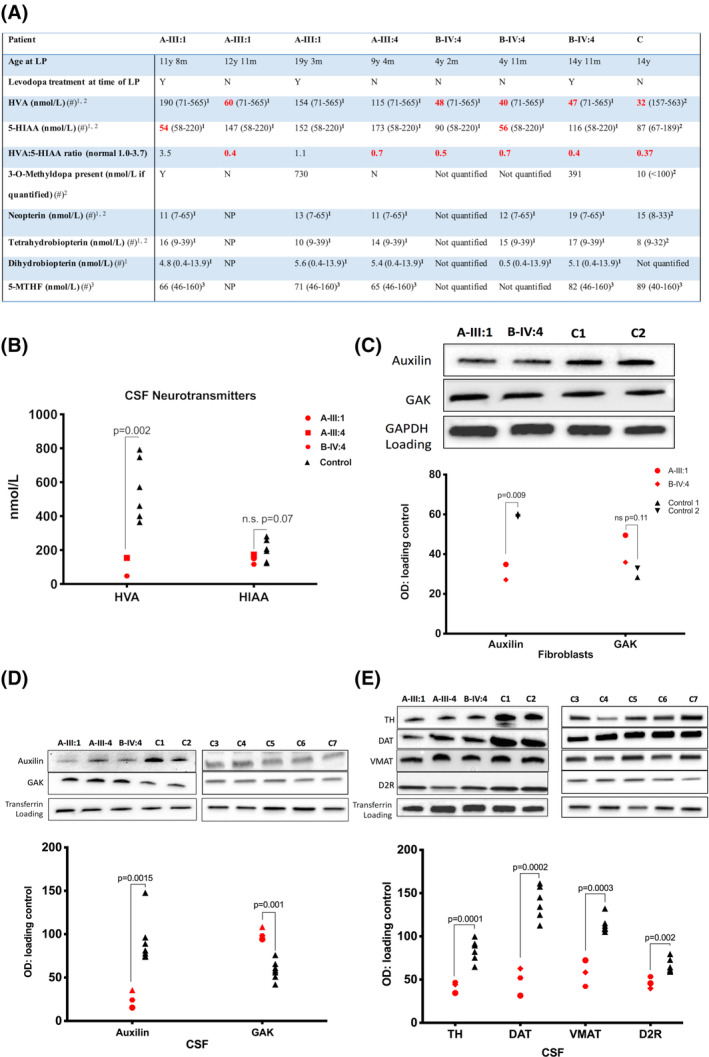
Results: We identified 6 patients harboring previously unreported, homozygous nonsense DNAJC6 mutations. All presented with neurodevelopmental delay in infancy, progressive parkinsonism, and neurological regression in childhood. 123 I-FP-CIT SPECT (DaTScan) was performed in 3 patients and demonstrated reduced or absent tracer uptake in the basal ganglia. CSF neurotransmitter analysis revealed an isolated reduction of homovanillic acid. Auxilin levels were significantly reduced in both patient fibroblasts and CSF. Cyclin G-associated kinase levels in CSF were significantly increased, whereas a number of presynaptic dopaminergic proteins were reduced.
Conclusions: DNAJC6 is an emerging cause of recessive juvenile parkinsonism-dystonia. DNAJC6 encodes the cochaperone protein auxilin, involved in CME of synaptic vesicles. The observed dopamine dyshomeostasis in patients is likely to be multifactorial, secondary to auxilin deficiency and/or neurodegeneration. Increased patient CSF cyclin G-associated kinase, in tandem with reduced auxilin levels, suggests a possible compensatory role of cyclin G-associated kinase, as observed in the auxilin knockout mouse. DNAJC6 parkinsonism-dystonia should be considered as a differential diagnosis for pediatric neurotransmitter disorders associated with low homovanillic acid levels. Future research in elucidating disease pathogenesis will aid the development of better treatments for this pharmacoresistant disorder. © 2020 The Authors. Movement Disorders published by Wiley Periodicals, Inc. on behalf of International Parkinson and Movement Disorder Society.
Keywords: DNAJC6; auxilin; dopamine; dystonia; parkinsonism.
© 2020 The Authors. Movement Disorders published by Wiley Periodicals, Inc. on behalf of International Parkinson and Movement Disorder Society.
Figures

References
-
- Kitada T, Asakawa S, Hattori N, et al. Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature 1998;392:605–608. - PubMed
-
- Valente EM, Abou‐Sleiman PM, Caputo V, et al. Hereditary early‐onset Parkinson's disease caused by mutations in PINK1. Science 2004;304:1158–1160. - PubMed
-
- Bonifati V, Rizzu P, van Baren MJ, et al. Mutations in the DJ‐1 gene associated with autosomal recessive early‐onset parkinsonism. Science 2003;299:256–259. - PubMed
-
- Ramirez A, Heimbach A, Gründemann J, et al. Hereditary parkinsonism with dementia is caused by mutations in ATP13A2, encoding a lysosomal type 5 P‐type ATPase. Nat Genet 2006;38:1184–1191. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
