

Phenotype expansion of heterozygous FOXC1 pathogenic variants toward involvement of congenital anomalies of the kidneys and urinary tract (CAKUT)
- PMID: 32475988
- PMCID: PMC8220407
- DOI: 10.1038/s41436-020-0844-z
Phenotype expansion of heterozygous FOXC1 pathogenic variants toward involvement of congenital anomalies of the kidneys and urinary tract (CAKUT)
Abstract
Purpose: Congenital anomalies of the kidney and urinary tract (CAKUT) are the most common cause of chronic kidney disease in childhood and adolescence. We aim to identify novel monogenic causes of CAKUT.
Methods: Exome sequencing was performed in 550 CAKUT-affected families.
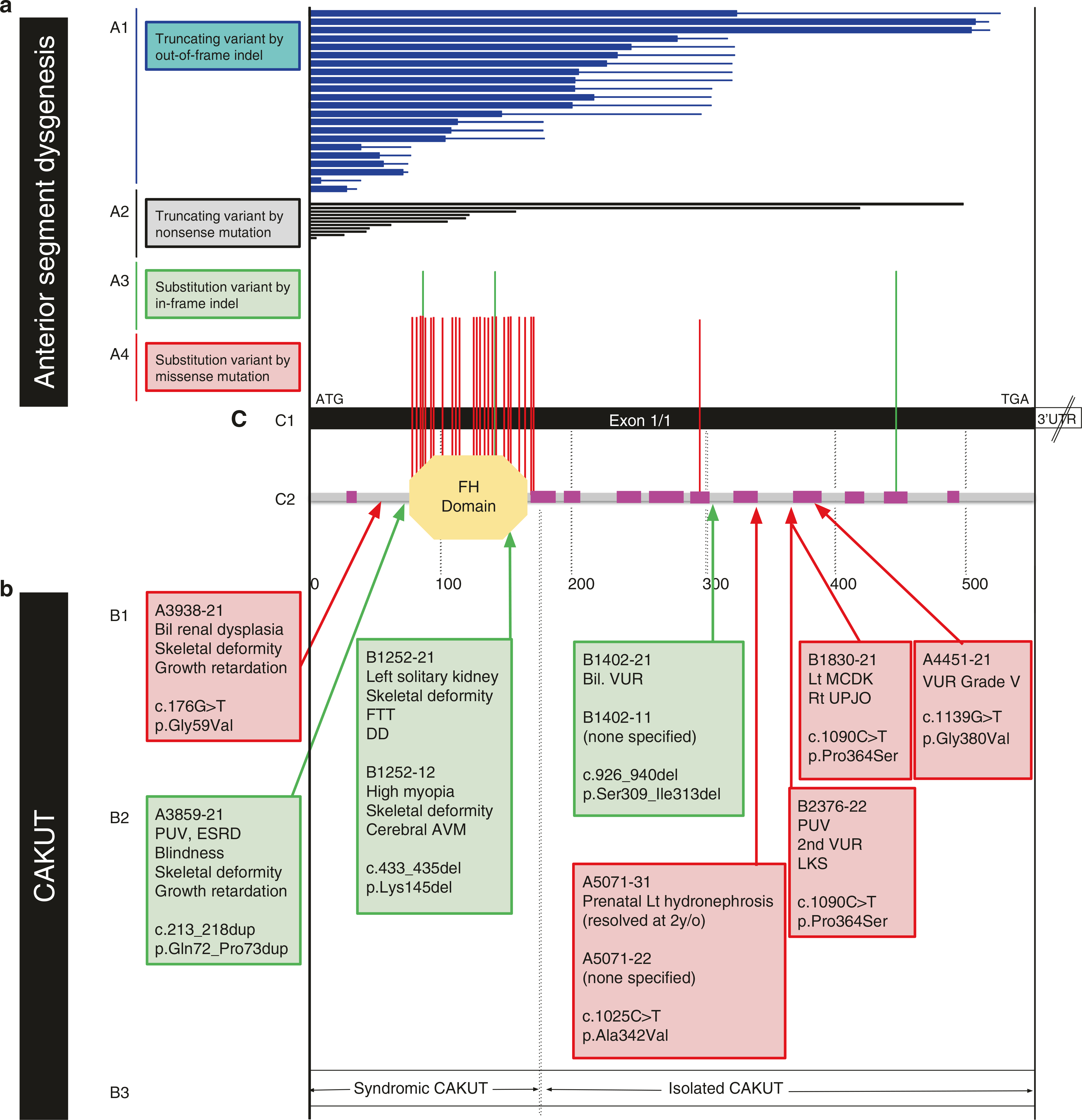
Results: We discovered seven FOXC1 heterozygous likely pathogenic variants within eight CAKUT families. These variants are either never reported, or present in <5 alleles in the gnomAD database with ~141,456 controls. FOXC1 is a causal gene for Axenfeld-Rieger syndrome type 3 and anterior segment dysgenesis 3. Pathogenic variants in FOXC1 have not been detected in patients with CAKUT yet. Interestingly, mouse models for Foxc1 show severe CAKUT phenotypes with incomplete penetrance and variable expressivity. The FOXC1 variants are enriched in the CAKUT cohort compared with the control. Genotype-phenotype correlations showed that Axenfeld-Rieger syndrome or anterior segment dysgenesis can be caused by both truncating and missense pathogenic variants, and the missense variants are located at the forkhead domain. In contrast, for CAKUT, there is no truncating pathogenic variant, and all variants except one are located outside the forkhead domain.
Conclusion: We thereby expanded the phenotype of FOXC1 pathogenic variants toward involvement of CAKUT, which can potentially be explained by allelism.
Keywords: congenital anomalies of the kidneys and urinary tract; exome sequencing.
Conflict of interest statement
DISCLOSURE
F.H. is a cofounder and S.A.C. member and holds stock in Goldfinch-Bio. All other authors declare that they have no competing financial interests.
Figures

References
-
- Chesnaye N, Bonthuis M, Schaefer F, et al. Demographics of paediatric renal replacement therapy in Europe: a report of the ESPN/ERA–EDTA registry. Pediatr Nephrol 2014;29:2403–2410. - PubMed
-
- Soliman NA, Ali RI, Ghobrial EE, Habib EI, Ziada AM. Pattern of clinical presentation of congenital anomalies of the kidney and urinary tract among infants and children. Nephrology. 2015;20:413–418. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
