

From GWAS to Function: Using Functional Genomics to Identify the Mechanisms Underlying Complex Diseases
- PMID: 32477401
- PMCID: PMC7237642
- DOI: 10.3389/fgene.2020.00424
From GWAS to Function: Using Functional Genomics to Identify the Mechanisms Underlying Complex Diseases
Abstract
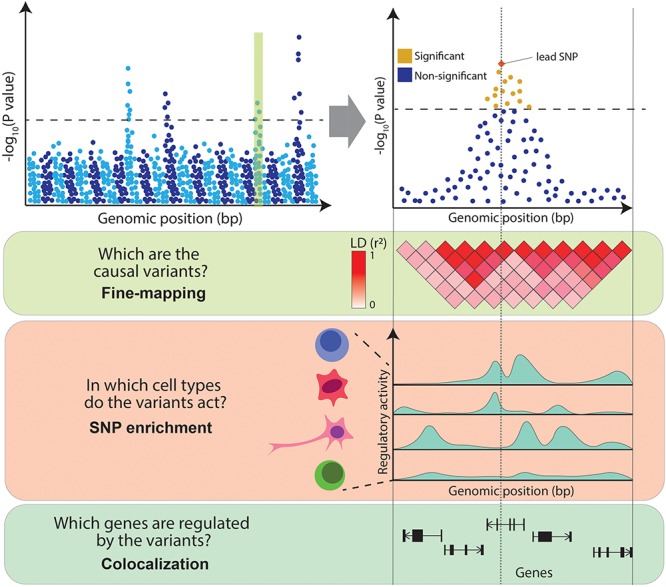
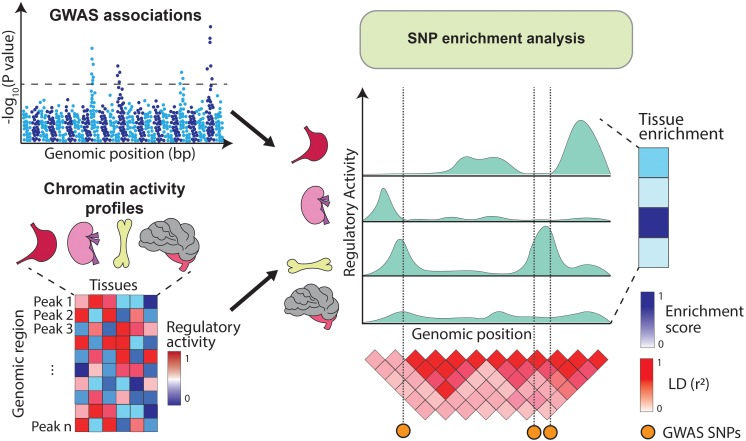
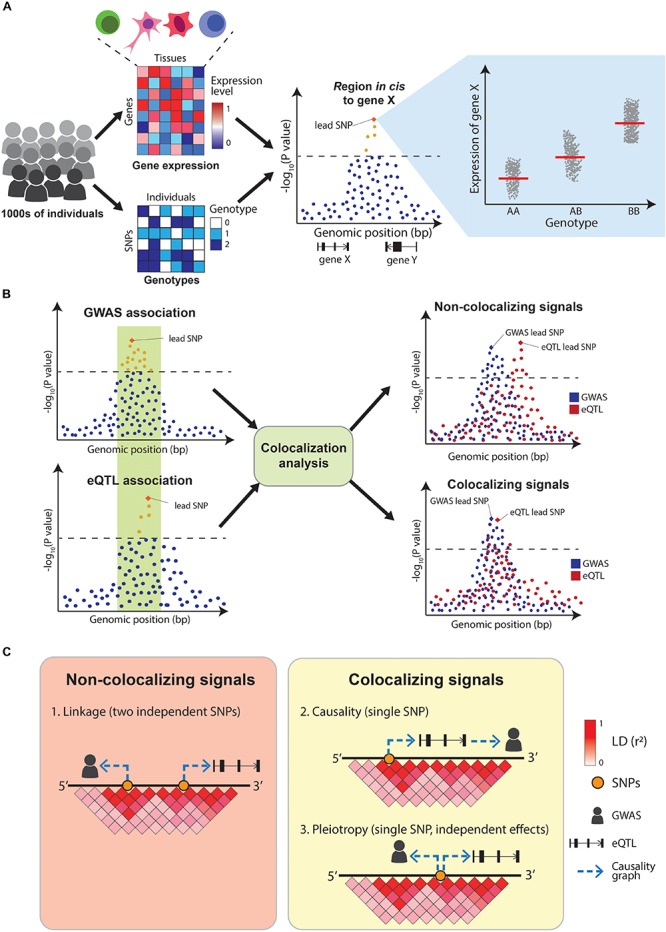
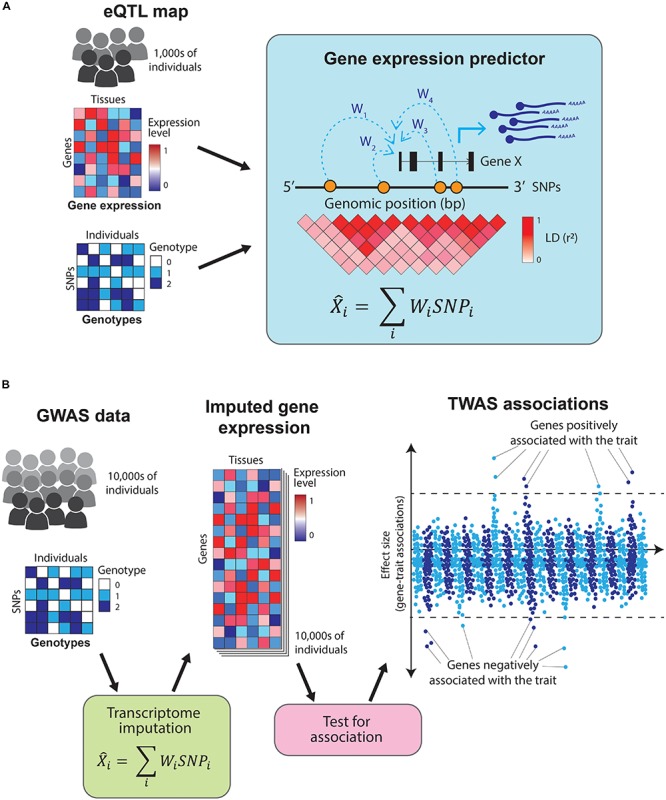
Genome-wide association studies (GWAS) have successfully mapped thousands of loci associated with complex traits. These associations could reveal the molecular mechanisms altered in common complex diseases and result in the identification of novel drug targets. However, GWAS have also left a number of outstanding questions. In particular, the majority of disease-associated loci lie in non-coding regions of the genome and, even though they are thought to play a role in gene expression regulation, it is unclear which genes they regulate and in which cell types or physiological contexts this regulation occurs. This has hindered the translation of GWAS findings into clinical interventions. In this review we summarize how these challenges have been addressed over the last decade, with a particular focus on the integration of GWAS results with functional genomics datasets. Firstly, we investigate how the tissues and cell types involved in diseases can be identified using methods that test for enrichment of GWAS variants in genomic annotations. Secondly, we explore how to find the genes regulated by GWAS loci using methods that test for colocalization of GWAS signals with molecular phenotypes such as quantitative trait loci (QTLs). Finally, we highlight potential future research avenues such as integrating GWAS results with single-cell sequencing read-outs, designing functionally informed polygenic risk scores (PRS), and validating disease associated genes using genetic engineering. These tools will be crucial to identify new drug targets for common complex diseases.
Keywords: GWAS; QTL; SNP enrichment; TWAS; colocalization analysis; eQTL; single-cell RNA seq.
Copyright © 2020 Cano-Gamez and Trynka.
Figures
References
-
- Amariuta T., Ishigaki K., Sugishita H., Ohta T., Matsuda K., Murakami Y., et al. (2020). In silico integration of thousands of epigenetic datasets into 707 cell type regulatory annotations improves the trans-ethnic portability of polygenic risk scores. bioRxiv [Preprint]
-
- Banovich N. E., Lan X., McVicker G., van de Geijn B., Degner J. F., Blischak J. D., et al. (2014). Methylation QTLs are associated with coordinated changes in transcription factor binding, histone modifications, and gene expression levels. PLoS Genet. 10:e1004663. 10.1371/journal.pgen.1004663 - DOI - PMC - PubMed
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources