

Desymmetrization of difluoromethylene groups by C-F bond activation
- PMID: 32480398
- PMCID: PMC10484566
- DOI: 10.1038/s41586-020-2399-1
Desymmetrization of difluoromethylene groups by C-F bond activation
Abstract
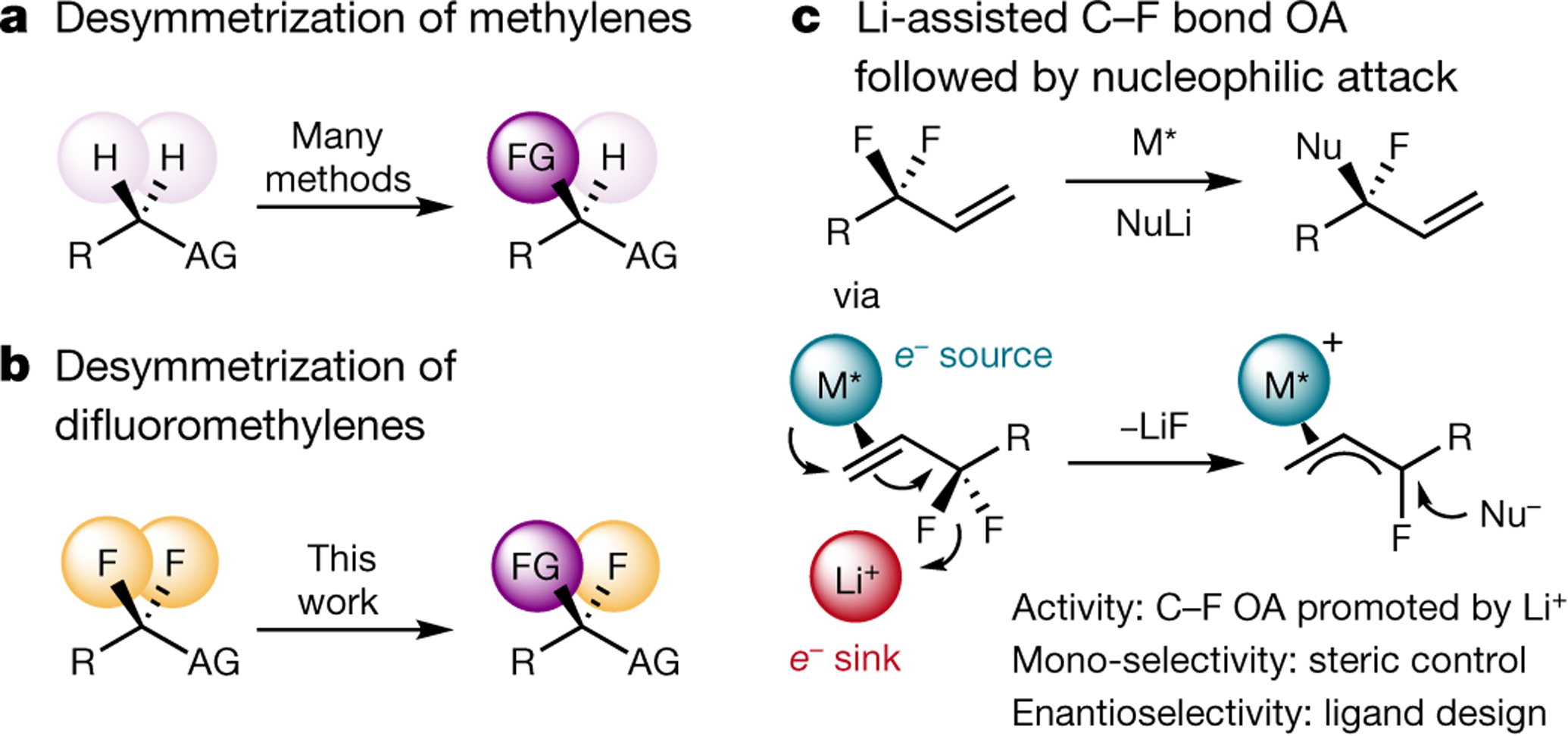
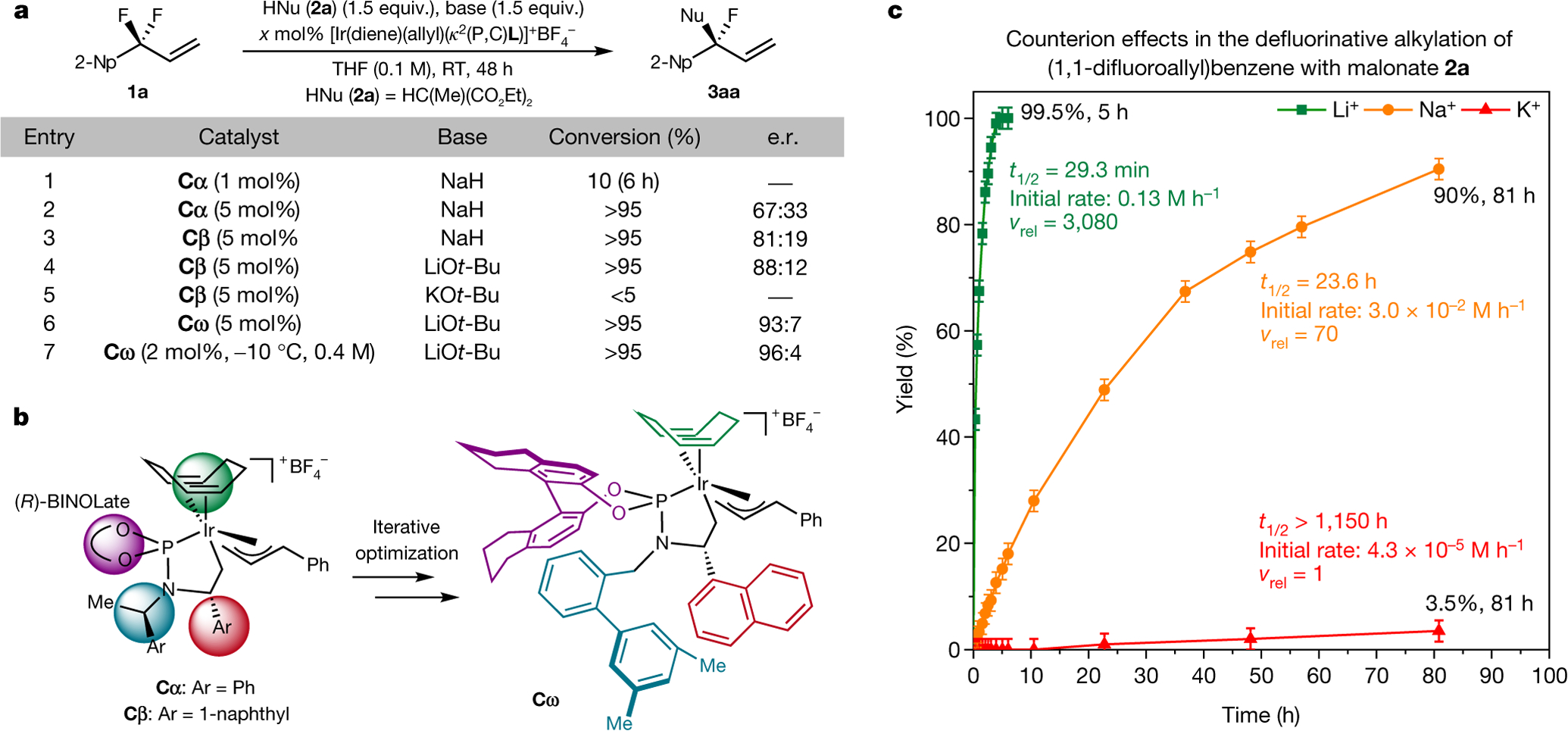
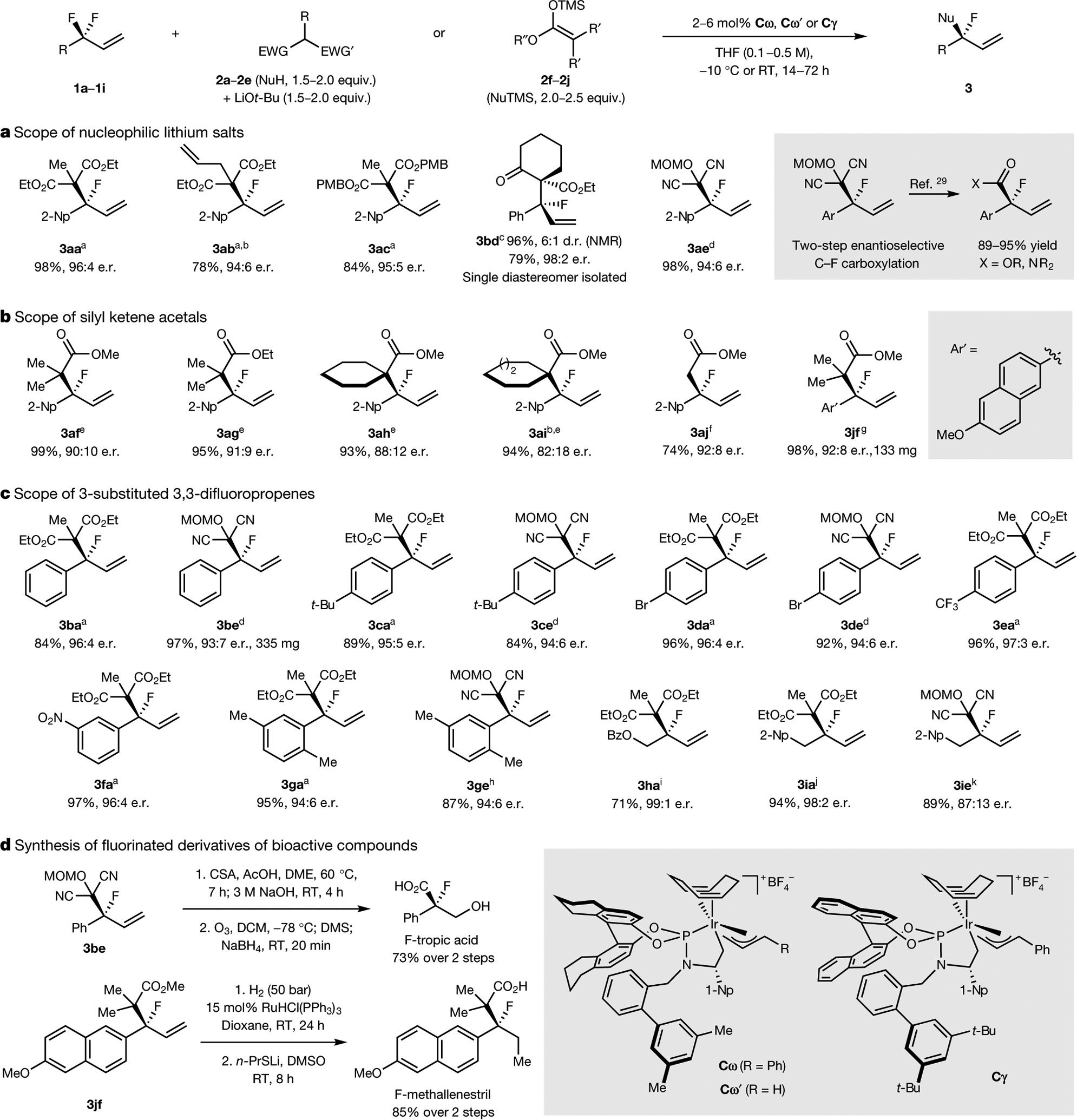
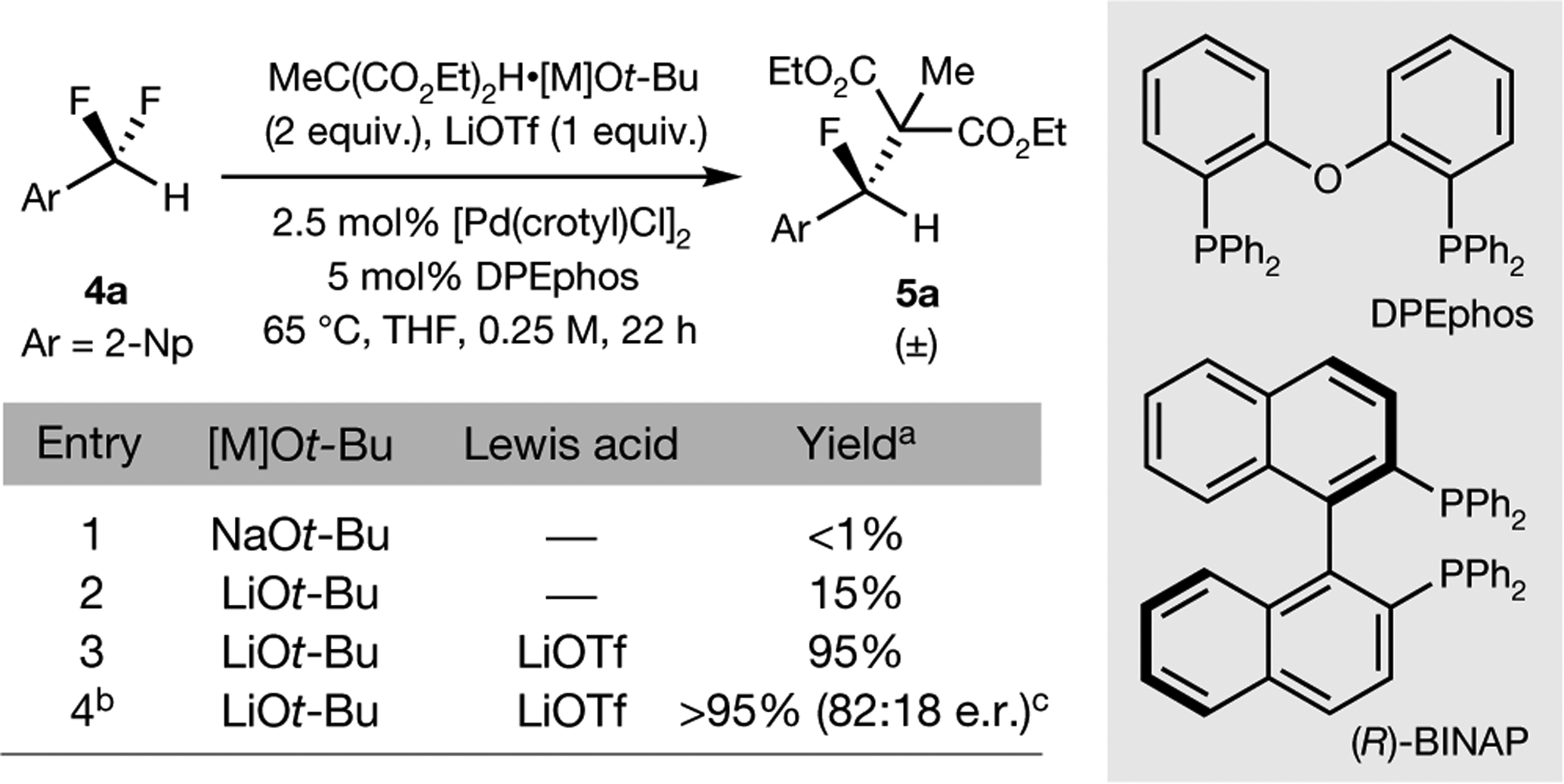
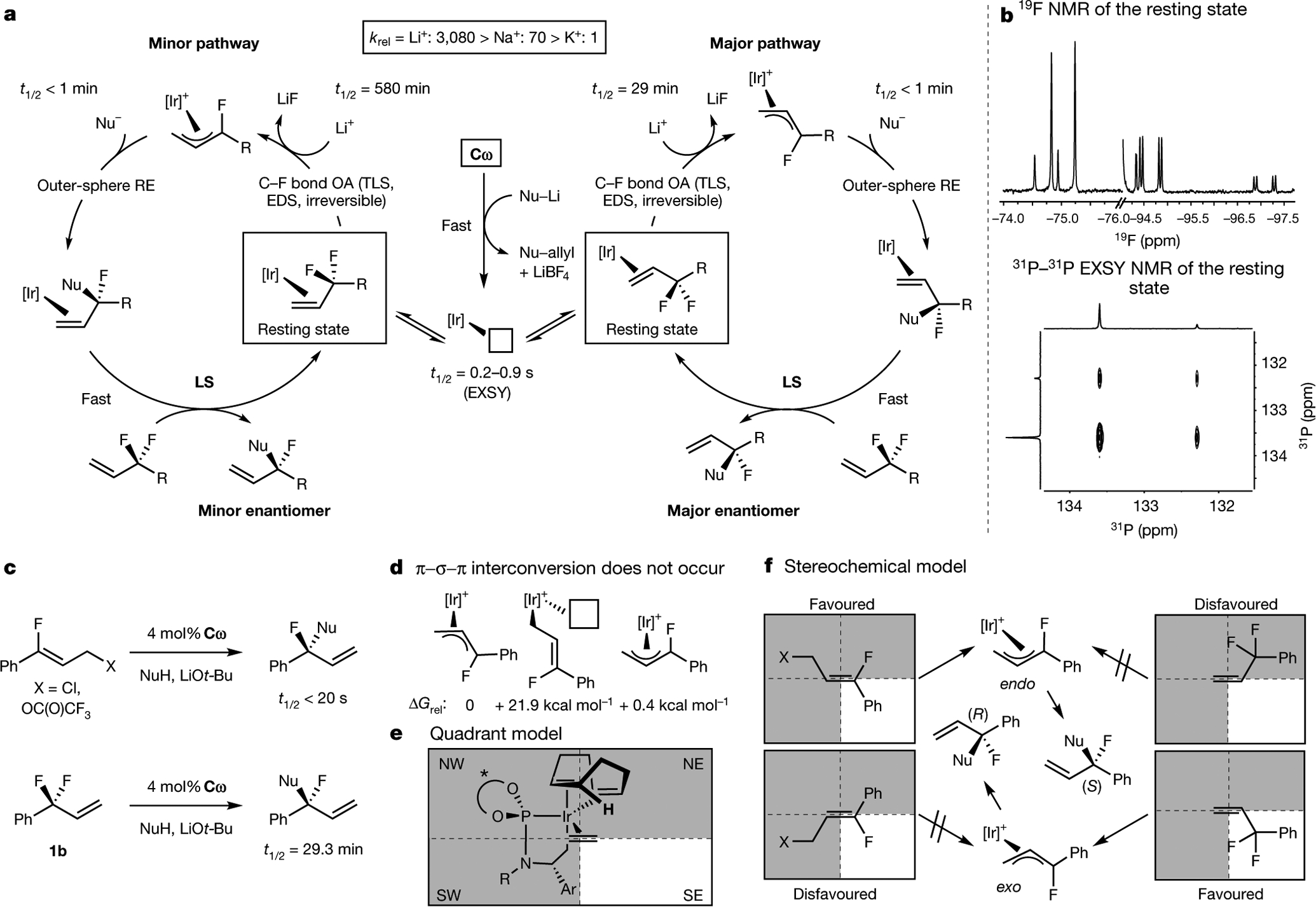
Tertiary stereogenic centres containing one fluorine atom are valuable for medicinal chemistry because they mimic common tertiary stereogenic centres containing one hydrogen atom, but they possess distinct charge distribution, lipophilicity, conformation and metabolic stability1-3. Although tertiary stereogenic centres containing one hydrogen atom are often set by enantioselective desymmetrization reactions at one of the two carbon-hydrogen (C-H) bonds of a methylene group, tertiary stereocentres containing fluorine have not yet been constructed by the analogous desymmetrization reaction at one of the two carbon-fluorine (C-F) bonds of a difluoromethylene group3. Fluorine atoms are similar in size to hydrogen atoms but have distinct electronic properties, causing C-F bonds to be exceptionally strong and geminal C-F bonds to strengthen one another4. Thus, exhaustive defluorination typically dominates over the selective replacement of a single C-F bond, hindering the development of the enantioselective substitution of one fluorine atom to form a stereogenic centre5,6. Here we report the catalytic, enantioselective activation of a single C-F bond in an allylic difluoromethylene group to provide a broad range of products containing a monofluorinated tertiary stereogenic centre. By combining a tailored chiral iridium phosphoramidite catalyst, which controls regioselectivity, chemoselectivity and enantioselectivity, with a fluorophilic activator, which assists the oxidative addition of the C-F bond, these reactions occur in high yield and selectivity. The design principles proposed in this work extend to palladium-catalysed benzylic substitution, demonstrating the generality of the approach.
Conflict of interest statement
Figures
References
-
- Purser S, Moore PR, Swallow S & Gouverneur V Fluorine in medicinal chemistry. Chem. Soc. Rev 37, 320–330 (2008). - PubMed
-
- Gillis EP, Eastman KJ, Hill MD, Donnelly DJ & Meanwell NA Applications of fluorine in medicinal chemistry. J. Med. Chem 58, 8315–8359 (2015). - PubMed
-
- O′Hagan D Understanding organofluorine chemistry. An introduction to the C–F bond. Chem. Soc. Rev 37, 308–319 (2008). - PubMed
-
- Stahl T, Klare HFT & Oestreich M Main-group Lewis acids for C–F bond activation. ACS Catal 3, 1578–1587 (2013).
