

Defective NADPH production in mitochondrial disease complex I causes inflammation and cell death
- PMID: 32483148
- PMCID: PMC7264245
- DOI: 10.1038/s41467-020-16423-1
Defective NADPH production in mitochondrial disease complex I causes inflammation and cell death
Abstract
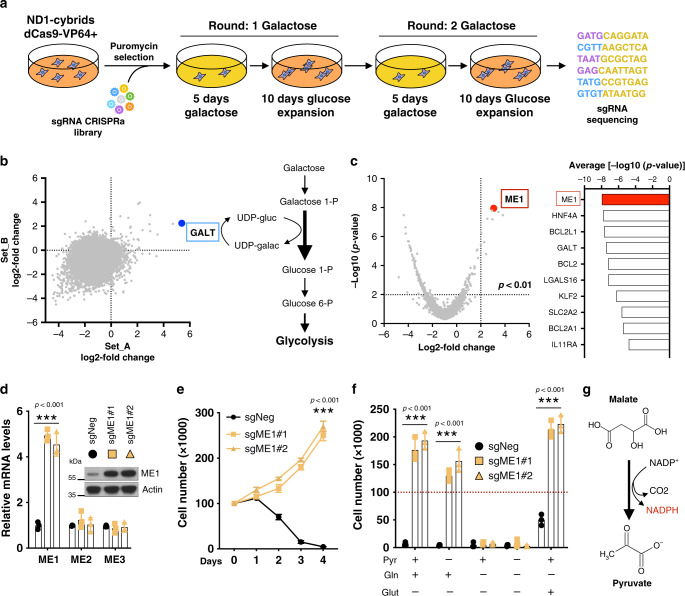
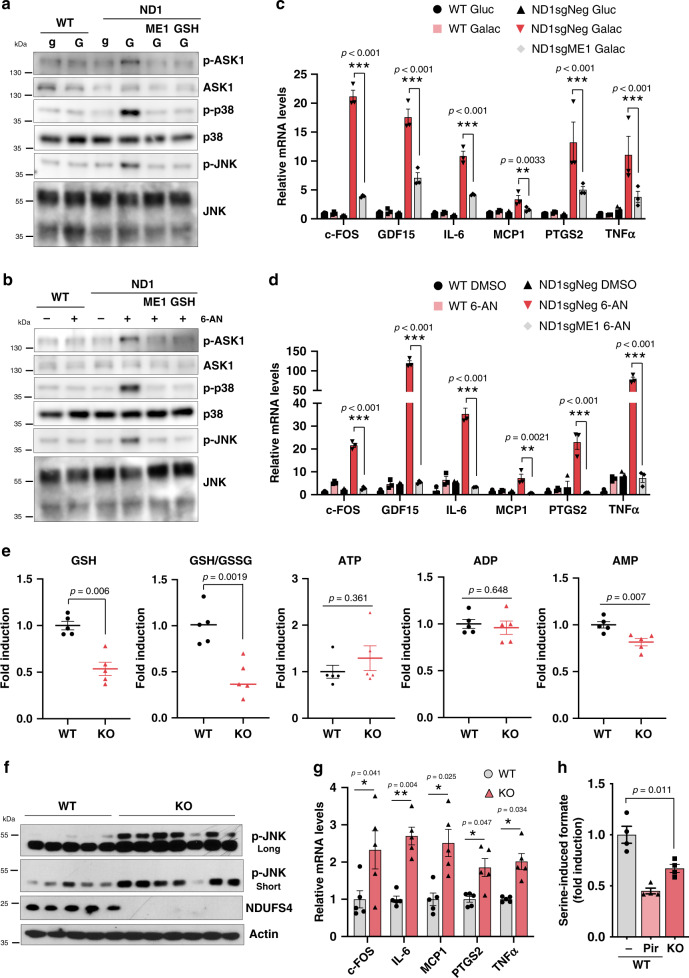
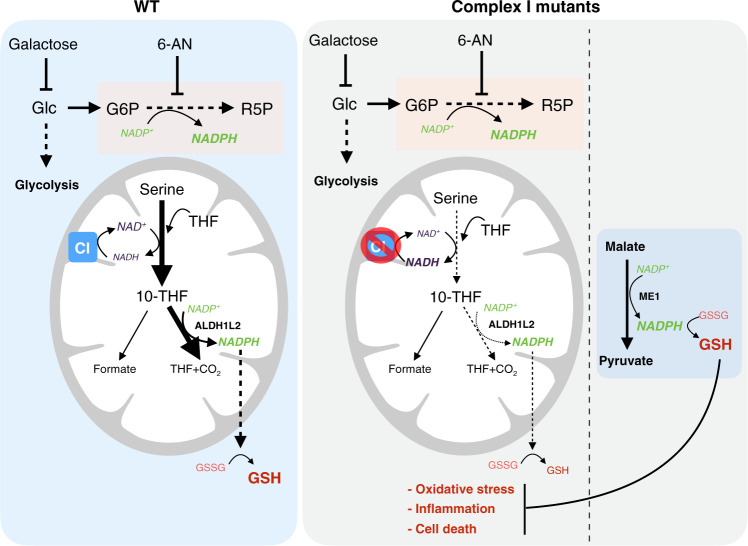
Electron transport chain (ETC) defects occurring from mitochondrial disease mutations compromise ATP synthesis and render cells vulnerable to nutrient and oxidative stress conditions. This bioenergetic failure is thought to underlie pathologies associated with mitochondrial diseases. However, the precise metabolic processes resulting from a defective mitochondrial ETC that compromise cell viability under stress conditions are not entirely understood. We design a whole genome gain-of-function CRISPR activation screen using human mitochondrial disease complex I (CI) mutant cells to identify genes whose increased function rescue glucose restriction-induced cell death. The top hit of the screen is the cytosolic Malic Enzyme (ME1), that is sufficient to enable survival and proliferation of CI mutant cells under nutrient stress conditions. Unexpectedly, this metabolic rescue is independent of increased ATP synthesis through glycolysis or oxidative phosphorylation, but dependent on ME1-produced NADPH and glutathione (GSH). Survival upon nutrient stress or pentose phosphate pathway (PPP) inhibition depends on compensatory NADPH production through the mitochondrial one-carbon metabolism that is severely compromised in CI mutant cells. Importantly, this defective CI-dependent decrease in mitochondrial NADPH production pathway or genetic ablation of SHMT2 causes strong increases in inflammatory cytokine signatures associated with redox dependent induction of ASK1 and activation of stress kinases p38 and JNK. These studies find that a major defect of CI deficiencies is decreased mitochondrial one-carbon NADPH production that is associated with increased inflammation and cell death.
Conflict of interest statement
The authors declare no competing interests.
Figures



References
Publication types
MeSH terms
Substances
Associated data
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
