

Selective Mediator dependence of cell-type-specifying transcription
- PMID: 32483291
- PMCID: PMC7610447
- DOI: 10.1038/s41588-020-0635-0
Selective Mediator dependence of cell-type-specifying transcription
Abstract
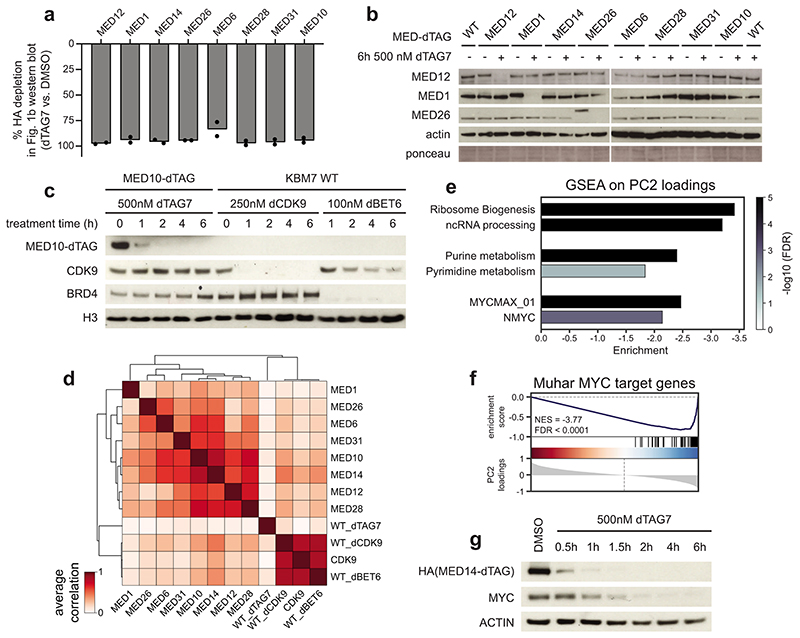
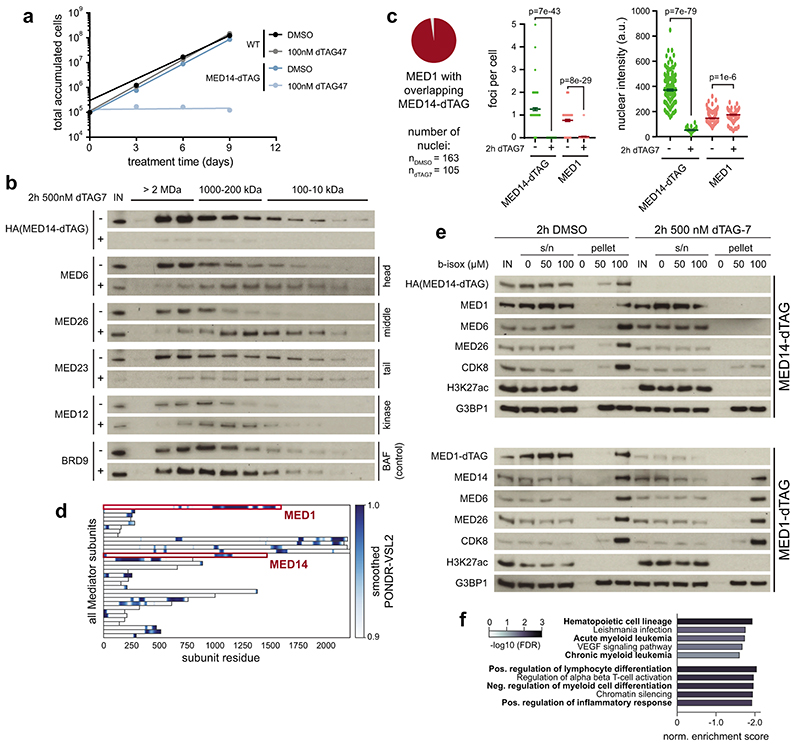
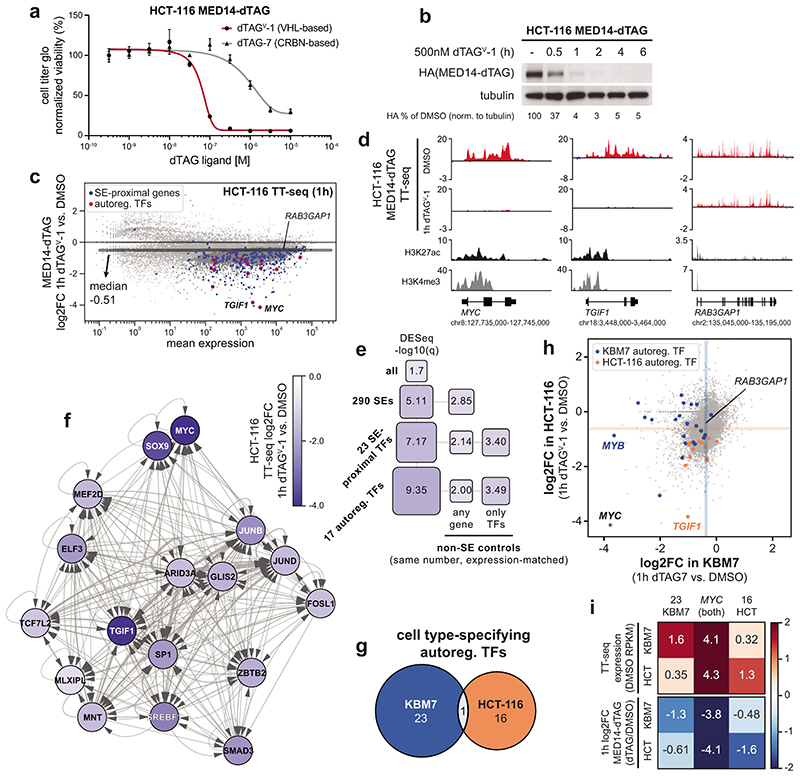
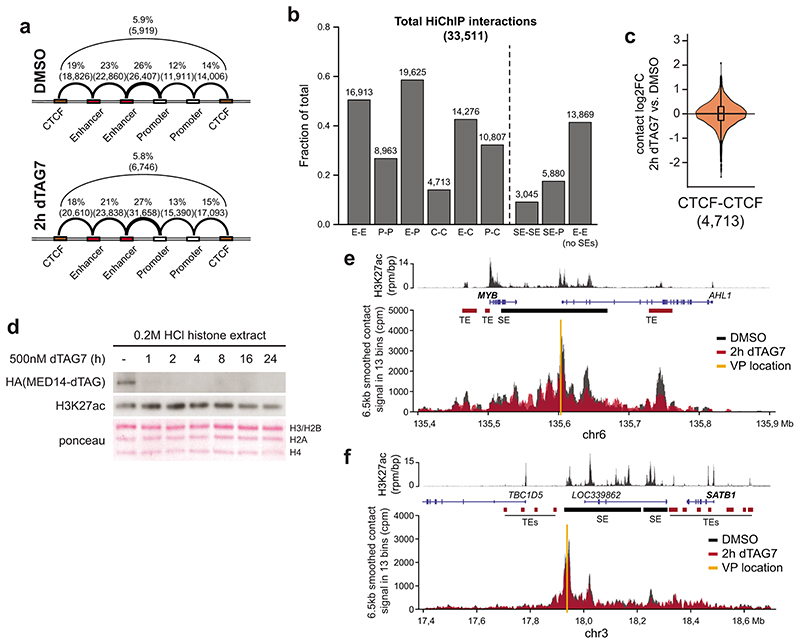
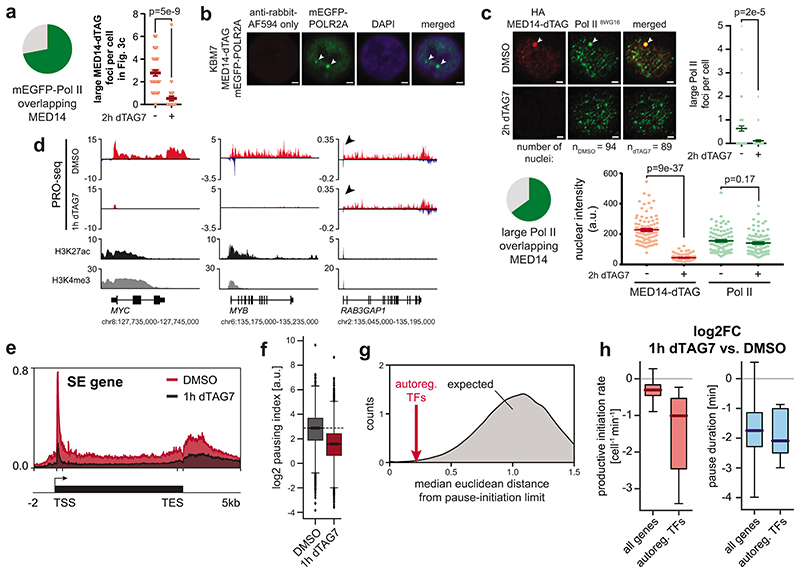
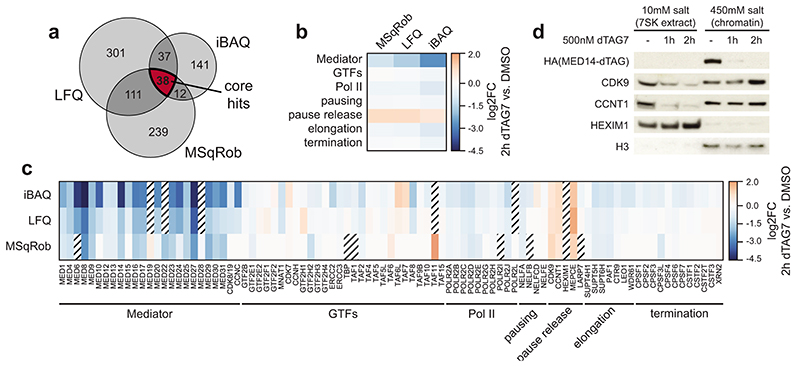
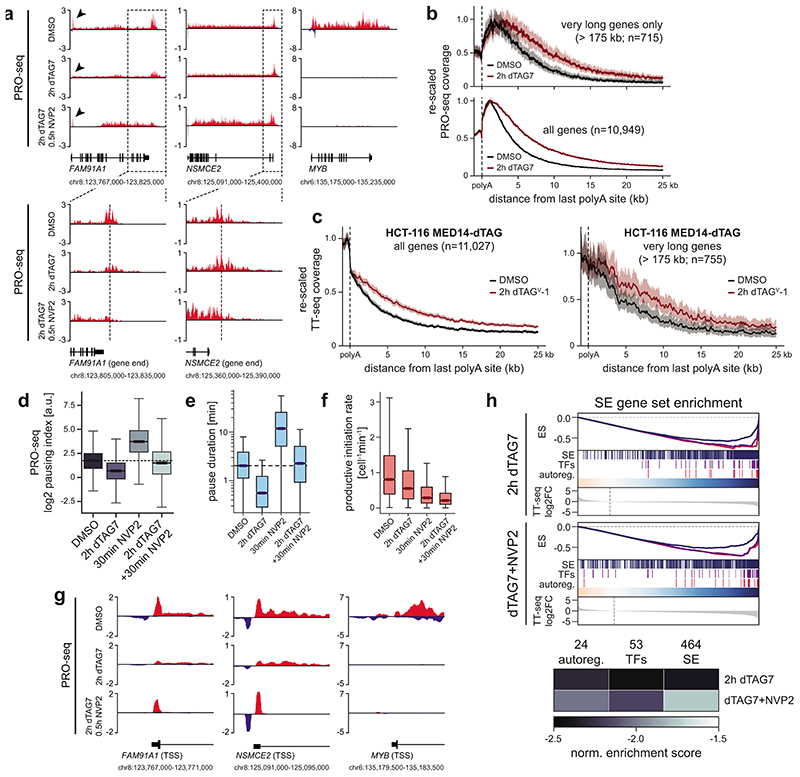
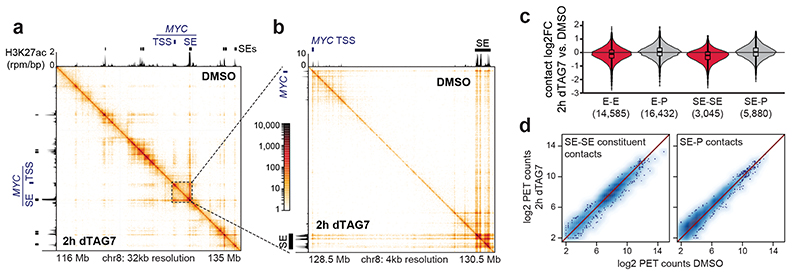
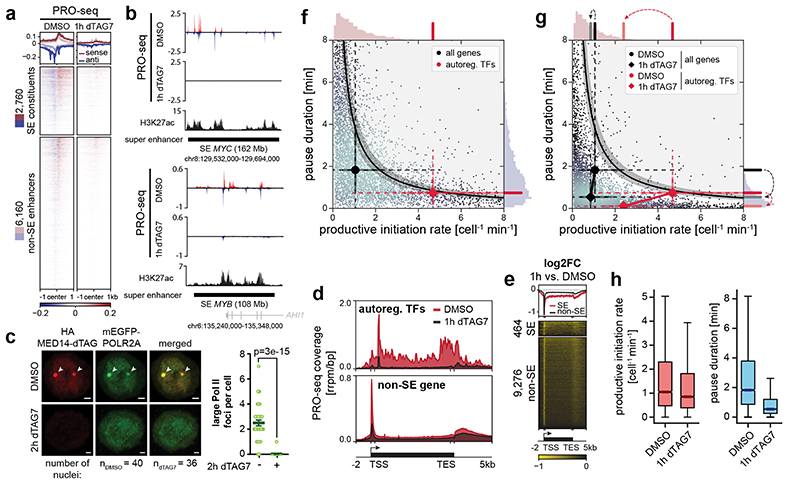
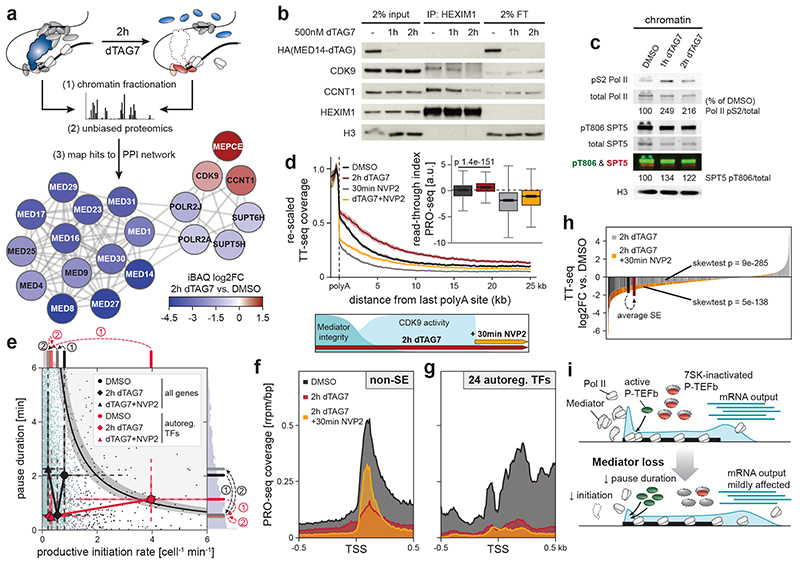
The Mediator complex directs signals from DNA-binding transcription factors to RNA polymerase II (Pol II). Despite this pivotal position, mechanistic understanding of Mediator in human cells remains incomplete. Here we quantified Mediator-controlled Pol II kinetics by coupling rapid subunit degradation with orthogonal experimental readouts. In agreement with a model of condensate-driven transcription initiation, large clusters of hypophosphorylated Pol II rapidly disassembled upon Mediator degradation. This was accompanied by a selective and pronounced disruption of cell-type-specifying transcriptional circuits, whose constituent genes featured exceptionally high rates of Pol II turnover. Notably, the transcriptional output of most other genes was largely unaffected by acute Mediator ablation. Maintenance of transcriptional activity at these genes was linked to an unexpected CDK9-dependent compensatory feedback loop that elevated Pol II pause release rates across the genome. Collectively, our work positions human Mediator as a globally acting coactivator that selectively safeguards the functionality of cell-type-specifying transcriptional networks.
Conflict of interest statement
G.E.W., J.E.B., and B.N. are inventors on patent applications related to the dTAG system (WO/2017/024318, WO/2017/024319, WO/2018/148443, WO/2018/148440). The dTAGV-1 molecule is the subject of a patent application filed by Dana-Farber Cancer Institute. N.S.G. is a Scientific Founder, member of the Scientific Advisory Board (SAB) and equity holder in C4 Therapeutics, Syros, Soltego, B2S, Gatekeeper and Petra Pharmaceuticals. The Gray lab receives or has received research funding from Novartis, Takeda, Astellas, Taiho, Janssen, Kinogen, Voroni, Her2llc, Deerfield and Sanofi. J.E.B. is now an executive and shareholder of Novartis AG, and has been a founder and shareholder of SHAPE (acquired by Medivir), Acetylon (acquired by Celgene), Tensha (acquired by Roche), Syros, Regenacy, and C4 Therapeutics.
Figures

References
-
- Kelleher RJ, 3rd, Flanagan PM, Kornberg RD. A novel mediator between activator proteins and the RNA polymerase II transcription apparatus. Cell. 1990;61:1209–15. - PubMed
-
- Kornberg RD. Mediator and the mechanism of transcriptional activation. Trends Biochem Sci. 2005;30:235–9. - PubMed
-
- Thompson CM, Koleske AJ, Chao DM, Young RA. A multisubunit complex associated with the RNA polymerase II CTD and TATA-binding protein in yeast. Cell. 1993;73:1361–75. - PubMed
-
- Kim YJ, Bjorklund S, Li Y, Sayre MH, Kornberg RD. A multiprotein mediator of transcriptional activation and its interaction with the C-terminal repeat domain of RNA polymerase II. Cell. 1994;77:599–608. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Miscellaneous
