

Review
doi: 10.1038/s41592-020-0848-2.
Epub 2020 Jun 1.
Macromolecular modeling and design in Rosetta: recent methods and frameworks
Affiliations
- PMID: 32483333
- PMCID: PMC7603796
- DOI: 10.1038/s41592-020-0848-2
Item in Clipboard
Review
Macromolecular modeling and design in Rosetta: recent methods and frameworks
Nat Methods.
2020 Jul.
Abstract
The Rosetta software for macromolecular modeling, docking and design is extensively used in laboratories worldwide. During two decades of development by a community of laboratories at more than 60 institutions, Rosetta has been continuously refactored and extended. Its advantages are its performance and interoperability between broad modeling capabilities. Here we review tools developed in the last 5 years, including over 80 methods. We discuss improvements to the score function, user interfaces and usability. Rosetta is available at http://www.rosettacommons.org.
Figures
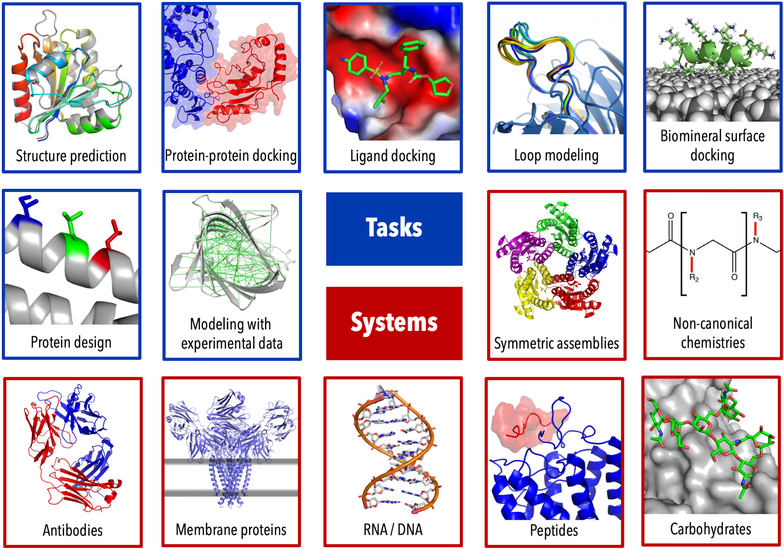
Some popular tasks that can be addressed in Rosetta (blue) and major systems that can be modeled (red). Note this is an incomplete list of Rosetta’s broad modeling capabilities.
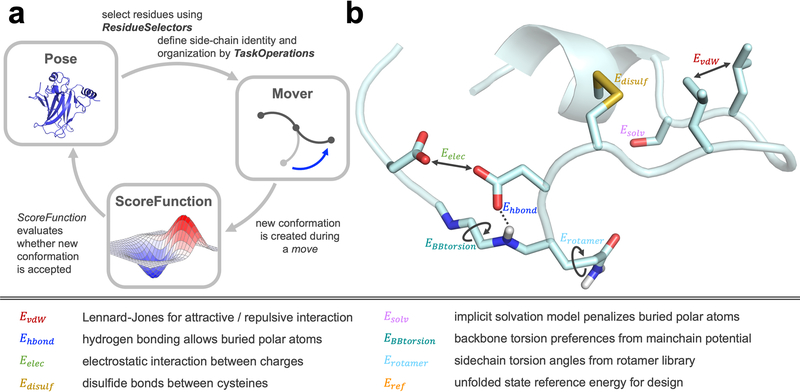
(A) Three main elements are required in a Rosetta protocol. The Pose is the biomolecule, such as a protein, RNA, DNA, small molecule, or glycan, in a specific conformation. Residues in the Pose can be selected via ResidueSelectors and the behavior for side-chain optimization or mutation can be defined by TaskOperations. Specific Movers then control how the conformation of the Pose is changed, and the new conformation is subsequently evaluated by a ScoreFunction. The Metropolis criterion decides whether the new conformation is accepted during sampling. Many independent sampling trajectories are generated, and the final models are evaluated based on the purpose of the protocol. (B) The score function consists of a weighted linear combination of various score terms, highlighted in the figure and described above.
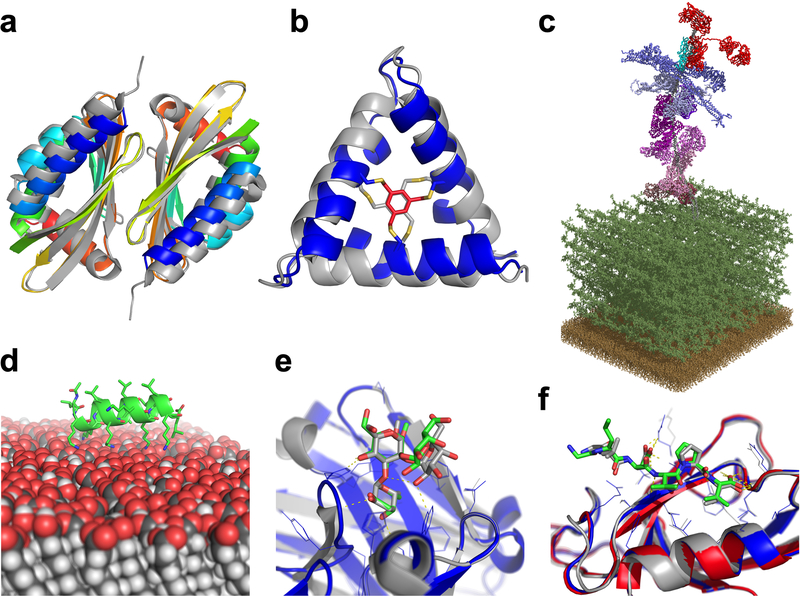
(A) Curved β-sheet design: overlay of the designed homo-dimeric curved β-sheet (dcs-E_4_dim_cav3) in rainbow and the crystal structure in gray (PDBID 5u35). The protein is designed de novo and features a curved β-sheet, a large pocket, and a homodimer interface. (B) Parametric design: overlay of the de novo designed macrocycle 3H1 in blue and the NMR structure in gray (PDBID 5v2g). This “CovCore” (covalent core) miniprotein is held together covalently by a hydrophobic cross-linker at its core (in red for the design and gray for the NMR structure). (C) PyTXMS: the interactome of M1 protein (virulence factor of Group A streptococcus) and 15 human plasma proteins on the surface of bacteria (peptidoglycan layer (dark green), and the membrane (brown)). This 1.8MDa structure contains over 200 chemical cross-links and is measured in a complex mixture of intact bacteria and human plasma. All models are provided by Rosetta: M1 protein (gray), IgG (red), four fibrinogens (dark to light blue), six albumins (dark to light pink), coagulation factor XIII A [F13A] (purple), C4bPa (cyan), haptoglobin [HP] (brown), and alpha-1-antitrypsin [SerpinA1] (plum). (D) RosettaSurface: model of an LK-α peptide (LKKLLKLLKKLLKL with a periodicity of 3.5 assuming a helical conformation) on a hydrophilic self-assembled monolayer surface. The peptide is unstructured in solution and assumes helical structure when on the surface, as experiments show. (E) RosettaCarbohydrate: flexible docking of a carbohydrate antigen to an antibody. The crystal structure is in gray (PDBID 1mfa) and the model in blue, with the carbohydrate in green. Antibody coordinates were taken from the PDB and glycan coordinates started from a randomized backbone conformation and rigid-body orientation. (F) PIPER-FlexPepDock: high-resolution model of a peptide-protein complex (model: blue; solved structure in gray, PDBID 1mfg). The model was generated from a peptide sequence (LDVPV, derived from the C-terminal tail of ErbB2R) and the unbound structure of the receptor (Erbin PDZ domain, PDBID 2h3l, colored in red).
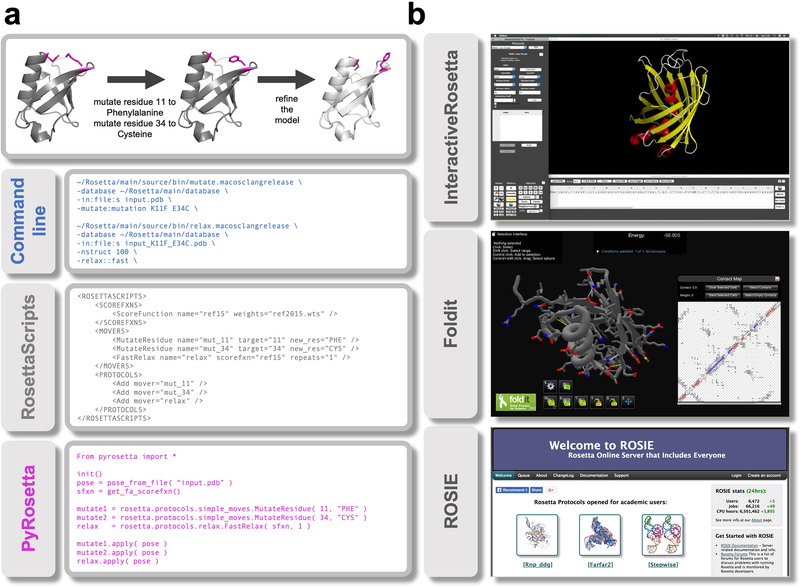
(A) Rosetta can be run from a terminal and offers three interfaces to the codebase. The top panel outlines the task to be accomplished: making two mutations in a protein and then refining the structure. The panels underneath show how this task can be accomplished in the different interfaces. The command line panel shows the executable, input files and options to run two specific applications. RosettaScripts is an XML-based scripting language that offers more flexibility by combining Movers and ScoreFunctions into a custom Protocol. PyRosetta offers direct access to the underlying code objects but requires knowledge of the codebase. (B) Point-and-click interfaces to the codebase. InteractiveRosetta is a graphical user-interface (GUI) to PyRosetta. It offers controls to the most popular protocols, file formats and options. Foldit is a videogame primarily used to crowd-source real-world scientific puzzles but can also be used on custom proteins of interest. It can run some popular applications via a game interface. ROSIE hosts a multitude of servers each executing a particular protocol. It currently includes servers for 21 Rosetta methods. [The InteractiveRosetta and Foldit panels were originally published in and under Creative Commons licenses that allows reproduction as is.]
In 2015, our community performed a complete overhaul of our documentation. Documentation is now hosted on a Gollum wiki, which is version controlled and easily editable by members of our community. Accessibility and ability to edit the documentation has drastically improved the user-experience of the software.
References
-
- Schrodinger - Biologics Design. at <https://www.schrodinger.com/science-articles/biologics-design>
-
- Molecular Operating Environment (MOE) | MOEsaic | PSILO. at <https://www.chemcomp.com/Products.htm>
-
- Ref. Dassault Systèmes BIOVIA, Discovery Studio Modeling Environment, Release 2017, San Diego: Dassault Systèmes, 2016. at <https://www.3dsbiovia.com/products/collaborative-science/biovia-discover...>
Publication types
MeSH terms
Substances
Grants and funding
- R01 GM099827/GM/NIGMS NIH HHS/United States
- R01 DK097376/DK/NIDDK NIH HHS/United States
- R01 GM117189/GM/NIGMS NIH HHS/United States
- T32 GM135141/GM/NIGMS NIH HHS/United States
- HHMI/Howard Hughes Medical Institute/United States
- RL1 CA133832/CA/NCI NIH HHS/United States
- R01 GM126299/GM/NIGMS NIH HHS/United States
- R01 GM117968/GM/NIGMS NIH HHS/United States
- F31 CA243353/CA/NCI NIH HHS/United States
- R21 GM102716/GM/NIGMS NIH HHS/United States
- R35 GM122517/GM/NIGMS NIH HHS/United States
- P30 CA006927/CA/NCI NIH HHS/United States
- F32 GM110899/GM/NIGMS NIH HHS/United States
- T32 GM007628/GM/NIGMS NIH HHS/United States
- P41 RR012408/RR/NCRR NIH HHS/United States
- R01 GM097207/GM/NIGMS NIH HHS/United States
- R01 GM099842/GM/NIGMS NIH HHS/United States
- R01 GM080403/GM/NIGMS NIH HHS/United States
- R01 GM092802/GM/NIGMS NIH HHS/United States
- R01 GM073151/GM/NIGMS NIH HHS/United States
- R35 GM125034/GM/NIGMS NIH HHS/United States
- R01 AI113867/AI/NIAID NIH HHS/United States
- R35 GM131923/GM/NIGMS NIH HHS/United States
- R01 GM127578/GM/NIGMS NIH HHS/United States
- R21 AI121799/AI/NIAID NIH HHS/United States
- R01 GM076324/GM/NIGMS NIH HHS/United States
- R01 GM088277/GM/NIGMS NIH HHS/United States
- R01 AI143997/AI/NIAID NIH HHS/United States
- R01 GM078221/GM/NIGMS NIH HHS/United States
- R01 GM123089/GM/NIGMS NIH HHS/United States
- R35 ES030443/ES/NIEHS NIH HHS/United States
- R01 GM132565/GM/NIGMS NIH HHS/United States
- P42 ES004699/ES/NIEHS NIH HHS/United States
- R35 GM122579/GM/NIGMS NIH HHS/United States
- T32 AI007244/AI/NIAID NIH HHS/United States
- R01 GM098101/GM/NIGMS NIH HHS/United States
- R01 GM099959/GM/NIGMS NIH HHS/United States
- F32 CA189246/CA/NCI NIH HHS/United States
- R01 GM110089/GM/NIGMS NIH HHS/United States
- F31 GM123616/GM/NIGMS NIH HHS/United States
- R01 HL122010/HL/NHLBI NIH HHS/United States
- 18POST34080422/AHA/American Heart Association-American Stroke Association/United States
- R01 GM084453/GM/NIGMS NIH HHS/United States
- R01 GM121487/GM/NIGMS NIH HHS/United States
- U19 AI117905/AI/NIAID NIH HHS/United States
- R00 GM120388/GM/NIGMS NIH HHS/United States
- UH2 CA203780/CA/NCI NIH HHS/United States
- R21 CA219847/CA/NCI NIH HHS/United States
- T32 GM008268/GM/NIGMS NIH HHS/United States
- R01 GM073960/GM/NIGMS NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
