

doi: 10.1038/s41587-020-0548-6.
PICRUSt2 for prediction of metagenome functions
Affiliations
- PMID: 32483366
- PMCID: PMC7365738
- DOI: 10.1038/s41587-020-0548-6
Item in Clipboard
PICRUSt2 for prediction of metagenome functions
Nat Biotechnol.
2020 Jun.
No abstract available
Figures
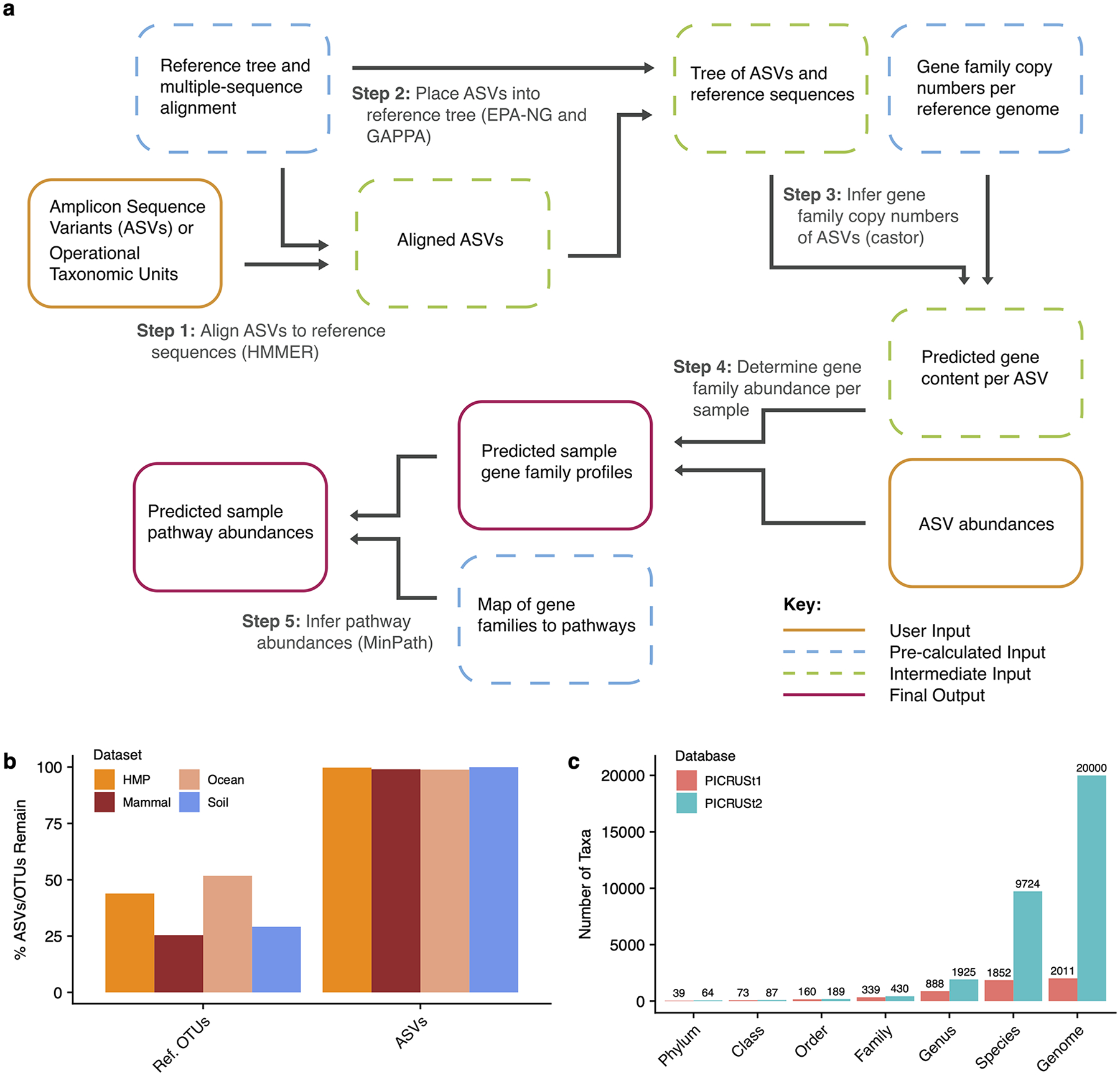
(a) The PICRUSt2 method consists of phylogenetic placement,
hidden-state-prediction and sample-wise gene and pathway abundance tabulation.
ASV sequences and abundances are taken as input, and gene family and pathway
abundances are output. All necessary reference tree and trait databases for the
default workflow are included in the PICRUSt2 implementation. (b) The default
PICRUSt1 pipeline restricted predictions to reference operational taxonomic
units (Ref. OTUs) in the Greengenes database. This requirement resulted in the
exclusion of many study sequences across four representative 16S rRNA gene
sequencing datasets. PICRUSt2 relaxes this requirement and is agnostic to
whether the input sequences are within a reference or not, which results in
almost all of the input amplicon sequence variants (ASVs) being retained in the
final output. (c) An increase in the taxonomic diversity in the default PICRUSt2
database is observed compared to PICRUSt1.
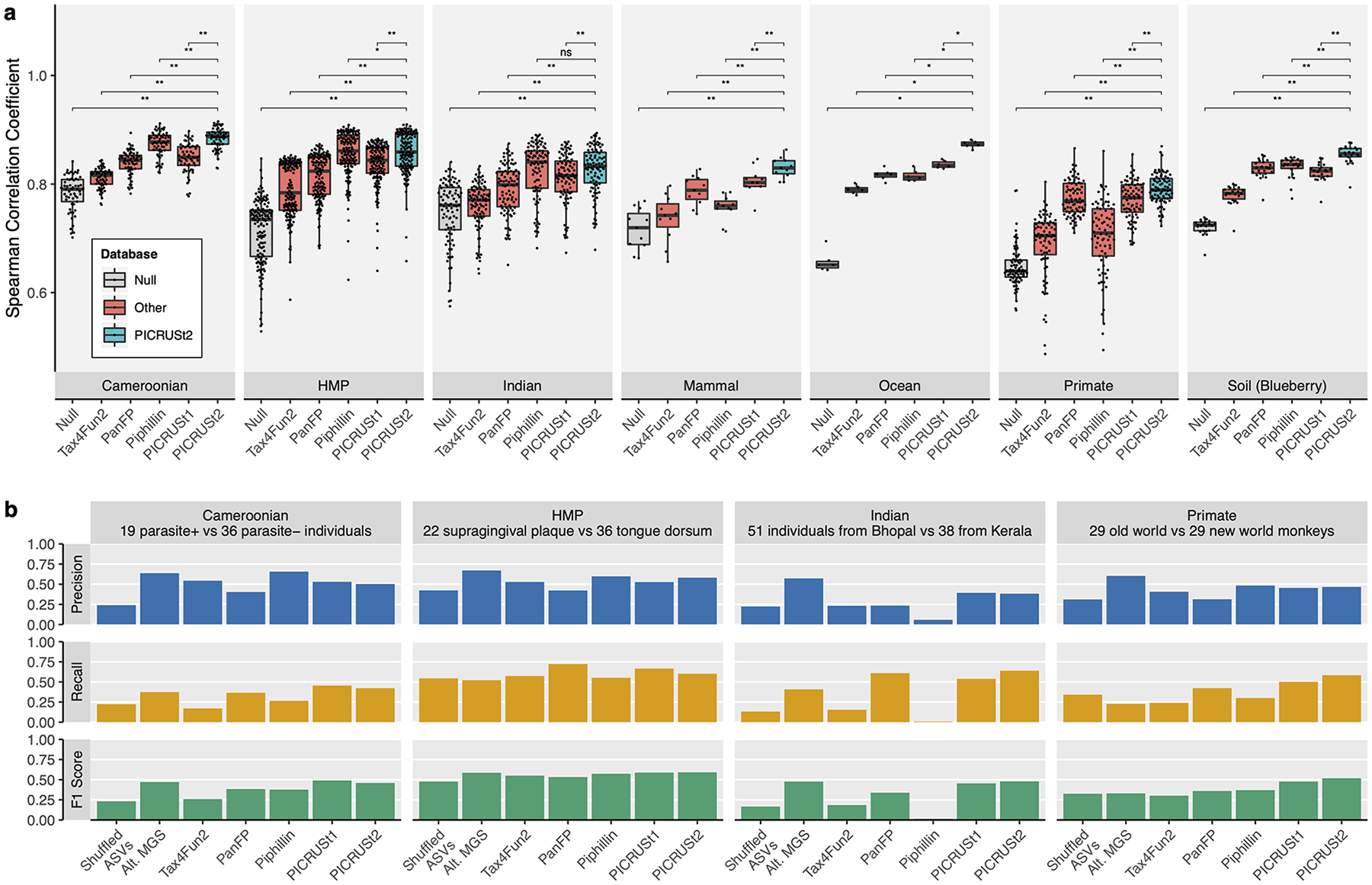
Validation of PICRUSt2 KEGG ortholog (KO) predictions comparing
metagenome prediction performance against gold-standard shotgun metagenomic
sequencing (MGS). (a) Boxplots of Spearman correlation coefficients observed in
stool samples from Cameroonian individuals (n=57), the human microbiome project
(HMP, n=137), stool samples from Indian individuals (n=91), non-human primate
stool samples (n=77), mammalian stool (n=8), ocean water (n=6), and blueberry
soil (n=22) datasets. The significance of paired-sample, two-tailed Wilcoxon
tests is indicated above each tested grouping (*, **, and ns correspond to P
< 0.05, P < 0.001, and not significant respectively). (b)
Comparison of significantly differentially abundant KOs between predicted
metagenomes and MGS. Precision, recall, and F1 score are reported for each
category compared to the MGS data. Precision corresponds to the proportion of
significant KOs for that category also significant in the MGS data. Recall
corresponds to the proportion of significant KOs in the MGS data also
significant for that category. The F1 score is the harmonic mean of these
metrics. The subsets of the four datasets compared are indicated above each
panel (the Cameroonian parasite is Entamoeba). Wilcoxon tests
were performed on the KO relative abundances after normalizing by the median
number of universal single-copy genes per sample. Significance was defined at a
false discovery rate < 0.05. The “Shuffled ASVs” category
corresponds to PICRUSt2 predictions with ASV labels shuffled per dataset. The
“Alt. MGS” category corresponds to an alternative MGS processing
pipeline with reads aligned to the KEGG database rather than the default HUMAnN2
pipeline.
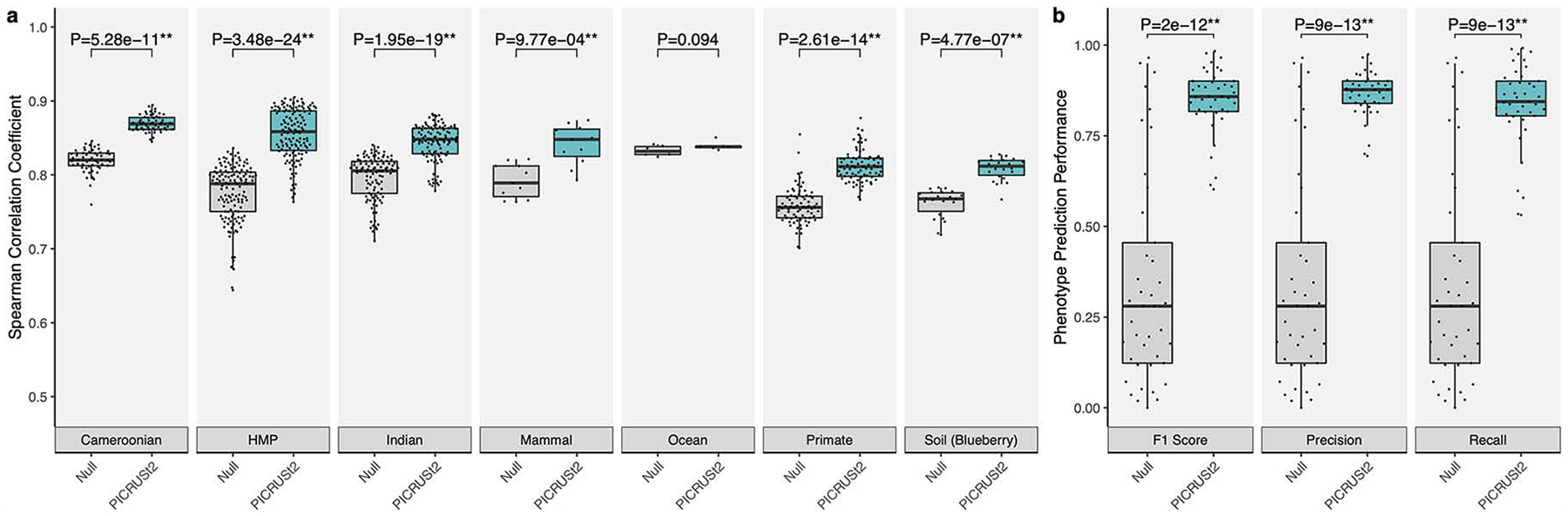
(a) Spearman correlation coefficients between PICRUSt2 predicted pathway
abundances and gold-standard metagenomic sequencing (MGS). Results are shown for
each validation dataset: stool from Cameroonian individuals, The Human
Microbiome Project (HMP), stool from Indian individuals, mammalian stool, ocean
water, non-human primate stool, and blueberry soil. These results are limited to
the 575 pathways that could potentially be identified by PICRUSt2 and HUMAnN2.
(b) Performance of binary phenotype predictions based on three metrics: F1
score, precision, and recall. Each point corresponds to one of the 41 phenotypes
tested. Predictions assessed here are based on holding out each genome
individually, predicting the phenotypes for that holdout genome, and comparing
the predicted and observed values. The null distribution in this case is based
on randomizing the phenotypes across the reference genomes and comparing to the
actual values, which results in the same output for all three metrics. The
P-values of paired-sample, two-tailed Wilcoxon tests is indicated above each
tested grouping (* and ** correspond to P < 0.05 and P < 0.001,
respectively). Note that in panel a the y-axis is truncated below 0.5 rather
than 0 to better visualize small differences between categories. The sample
sizes in panel a are 57 (Cameroonian), 137 (HMP), 91 (Indian), 8 (mammal), 6
(ocean), 77 (primate), and 22 (soil).
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
