

Comprehensive genomic diagnosis of inherited retinal and optical nerve disorders reveals hidden syndromes and personalized therapeutic options
- PMID: 32483926
- PMCID: PMC7754416
- DOI: 10.1111/aos.14479
Comprehensive genomic diagnosis of inherited retinal and optical nerve disorders reveals hidden syndromes and personalized therapeutic options
Abstract
Purpose: In the era of precision medicine, genomic characterization of blind patients is critical. Here, we evaluate the effects of comprehensive genetic analysis on the etiologic diagnosis of potentially hereditary vision loss and its impact on clinical management.
Methods: We studied 100 non-syndromic and syndromic Spanish patients with a clinical diagnosis of blindness caused by alterations on the retina, choroid, vitreous and/or optic nerve. We used a next-generation sequencing (NGS) panel (OFTALMOgenics™), developed and validated within this study, including up to 362 genes previously associated with these conditions.
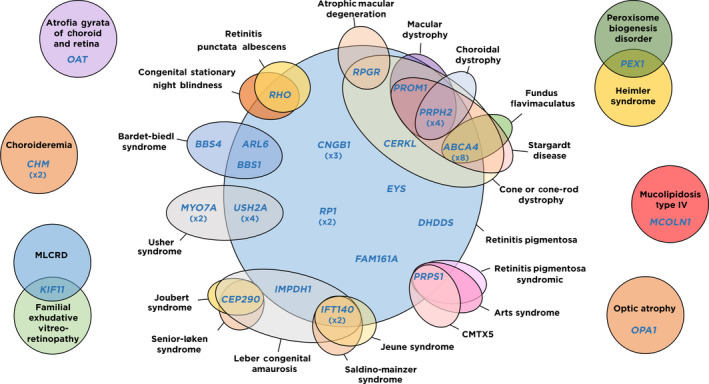
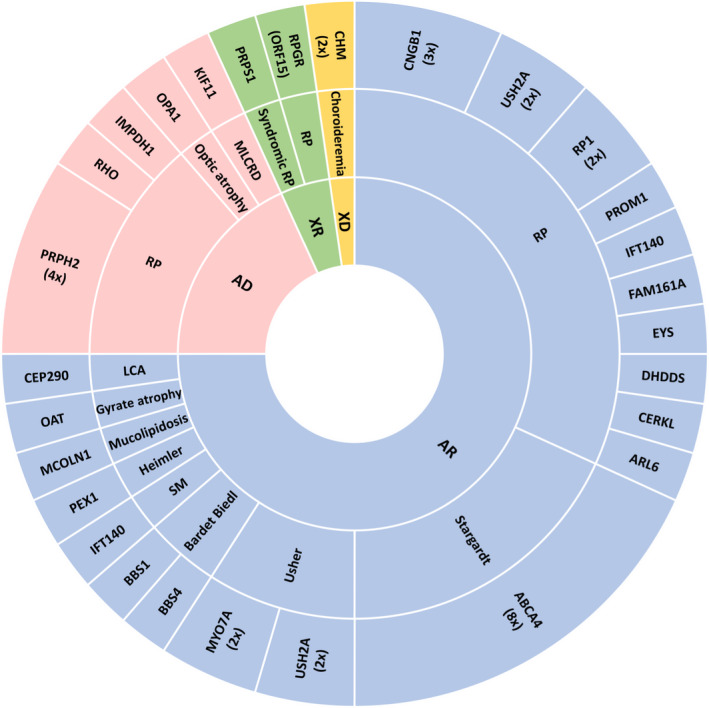
Results: We identified the genetic cause of blindness in 45% of patients (45/100). A total of 28.9% of genetically diagnosed cases (13/45) were syndromic and, of those, in 30.8% (4/13) extraophthalmic features had been overlooked and/or not related to visual impairment before genetic testing, including cases with Mainzer-Saldino, Bardet-Biedl, mucolipidosis and MLCRD syndromes. In two additional cases-syndromic blindness had been proposed before, but not specifically diagnosed, and one patient with Heimler syndrome had been misdiagnosed as an Usher case before testing. 33.3% of the genetically diagnosed patients (15/45) had causative variants in genes targeted by clinical trials exploring the curative potential of gene therapy approaches.
Conclusion: Comprehensive genomic testing provided clinically relevant insights in a large proportion of blind patients, identifying potential therapeutic opportunities or previously undiagnosed syndromes in 42.2% of the genetically diagnosed cases (19/45).
Keywords: genomic diagnostics; hereditary; inherited retinal dystrophies; next-generation sequencing; panel sequencing; precision ophthalmology; vision loss.
© 2020 The Authors. Acta Ophthalmologica published by John Wiley & Sons Ltd on behalf of Acta Ophthalmologica Scandinavica Foundation.
Figures
References
-
- Bamshad MJ, Ng SB, Bigham AW, Tabor HK, Emond MJ, Nickerson DA & Shendure J (2011): Exome sequencing as a tool for Mendelian disease gene discovery. Nat Rev Genet 12: 745–755. - PubMed
-
- Biesecker LG & Green RC (2014): Diagnostic clinical genome and exome sequencing. N Engl J Med 370: 2418–2425. - PubMed
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
