

Synaptic restoration by cAMP/PKA drives activity-dependent neuroprotection to motoneurons in ALS
- PMID: 32484501
- PMCID: PMC7398175
- DOI: 10.1084/jem.20191734
Synaptic restoration by cAMP/PKA drives activity-dependent neuroprotection to motoneurons in ALS
Abstract
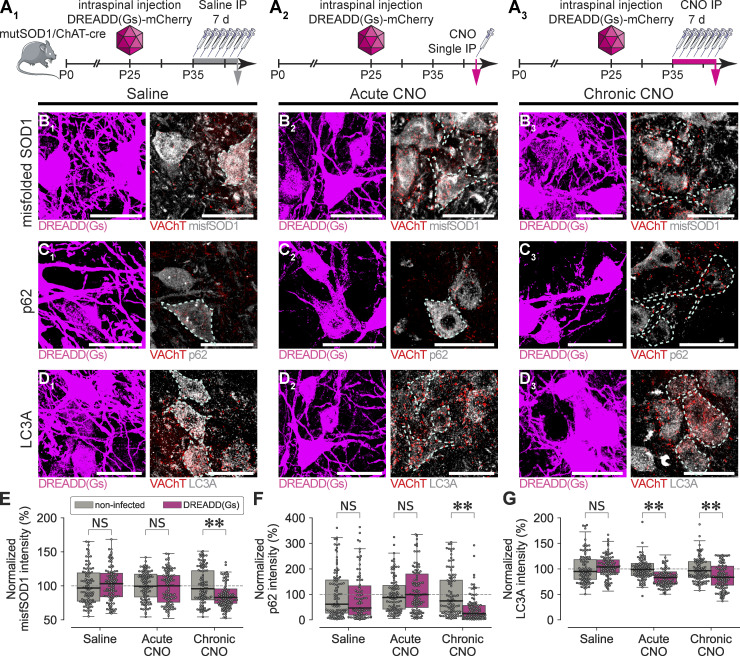
Excessive excitation is hypothesized to cause motoneuron (MN) degeneration in amyotrophic lateral sclerosis (ALS), but actual proof of hyperexcitation in vivo is missing, and trials based on this concept have failed. We demonstrate, by in vivo single-MN electrophysiology, that, contrary to expectations, excitatory responses evoked by sensory and brainstem inputs are reduced in MNs of presymptomatic mutSOD1 mice. This impairment correlates with disrupted postsynaptic clustering of Homer1b, Shank, and AMPAR subunits. Synaptic restoration can be achieved by activation of the cAMP/PKA pathway, by either intracellular injection of cAMP or DREADD-Gs stimulation. Furthermore, we reveal, through independent control of signaling and excitability allowed by multiplexed DREADD/PSAM chemogenetics, that PKA-induced restoration of synapses triggers an excitation-dependent decrease in misfolded SOD1 burden and autophagy overload. In turn, increased MN excitability contributes to restoring synaptic structures. Thus, the decrease of excitation to MN is an early but reversible event in ALS. Failure of the postsynaptic site, rather than hyperexcitation, drives disease pathobiochemistry.
© 2020 Bączyk et al.
Conflict of interest statement
Disclosures: The authors declare no competing interests exist.
Figures














References
-
- Aldrin-Kirk P., Heuer A., Wang G., Mattsson B., Lundblad M., Parmar M., and Björklund T.. 2016. DREADD Modulation of Transplanted DA Neurons Reveals a Novel Parkinsonian Dyskinesia Mechanism Mediated by the Serotonin 5-HT6 Receptor. Neuron. 90:955–968. 10.1016/j.neuron.2016.04.017 - DOI - PMC - PubMed
-
- Alvarez F.J., Titus-Mitchell H.E., Bullinger K.L., Kraszpulski M., Nardelli P., and Cope T.C.. 2011. Permanent central synaptic disconnection of proprioceptors after nerve injury and regeneration. I. Loss of VGLUT1/IA synapses on motoneurons. J. Neurophysiol. 106:2450–2470. 10.1152/jn.01095.2010 - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
