

BET bromodomain inhibition attenuates cardiac phenotype in myocyte-specific lamin A/C-deficient mice
- PMID: 32484798
- PMCID: PMC7456228
- DOI: 10.1172/JCI135922
BET bromodomain inhibition attenuates cardiac phenotype in myocyte-specific lamin A/C-deficient mice
Abstract
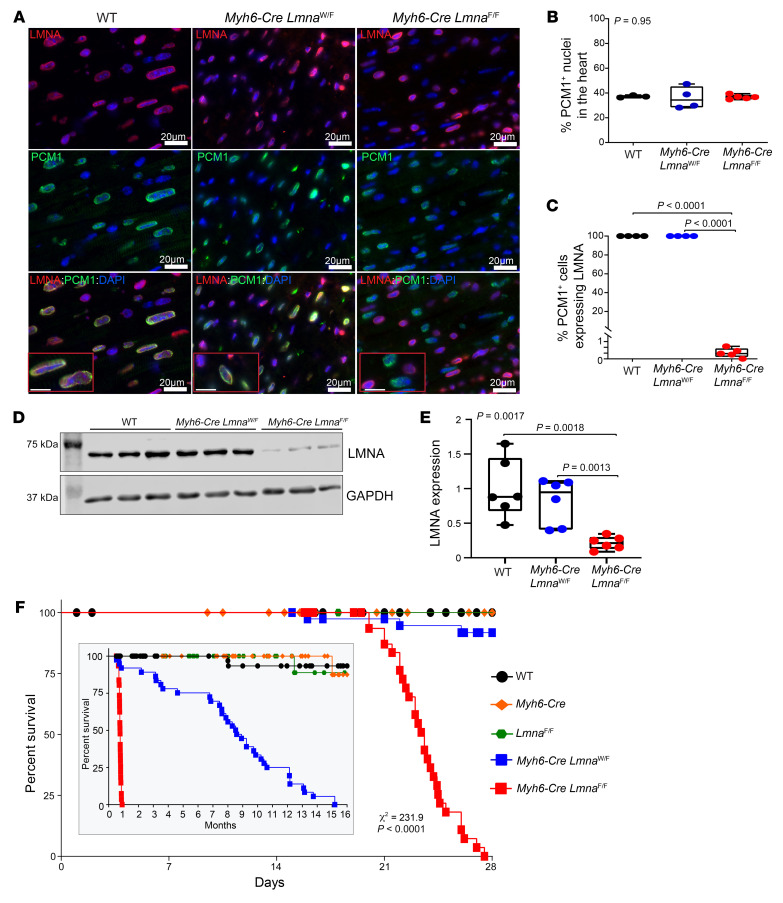
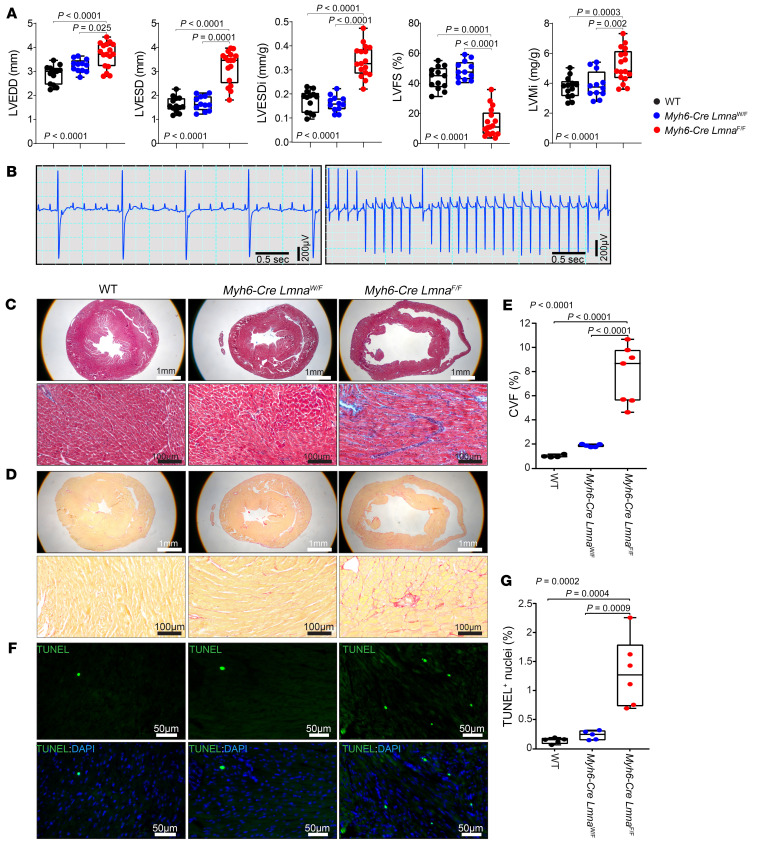
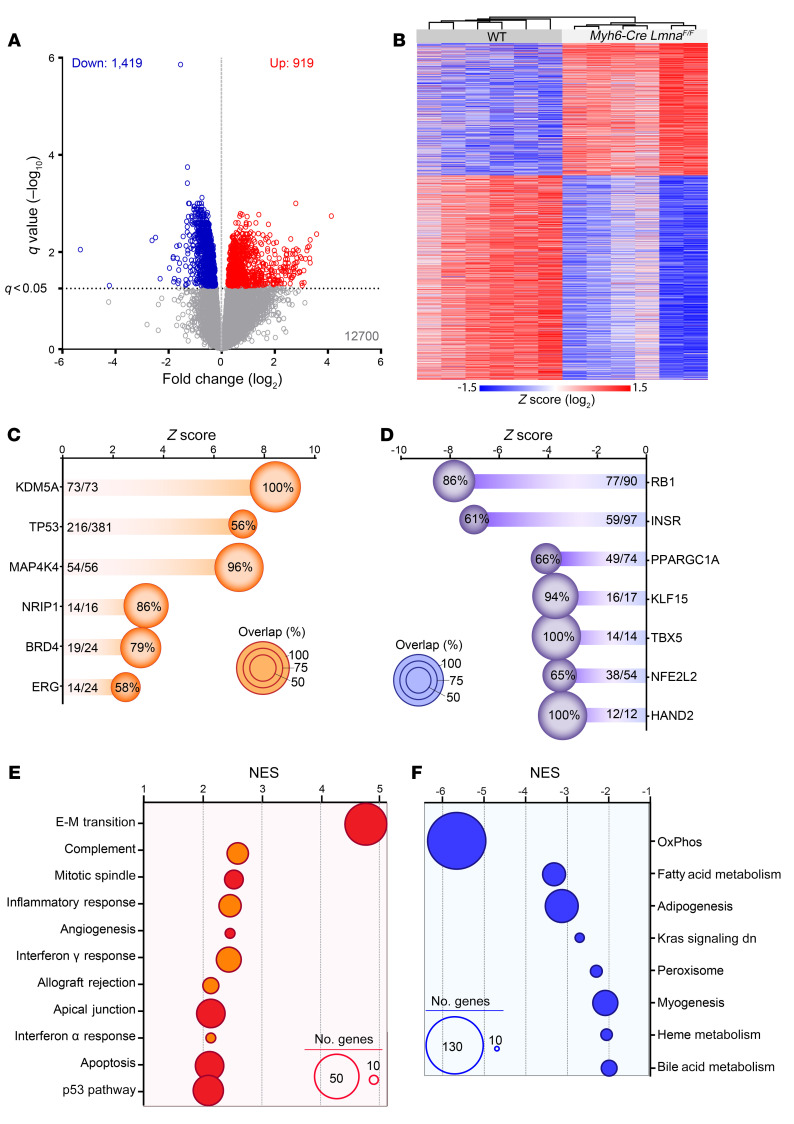
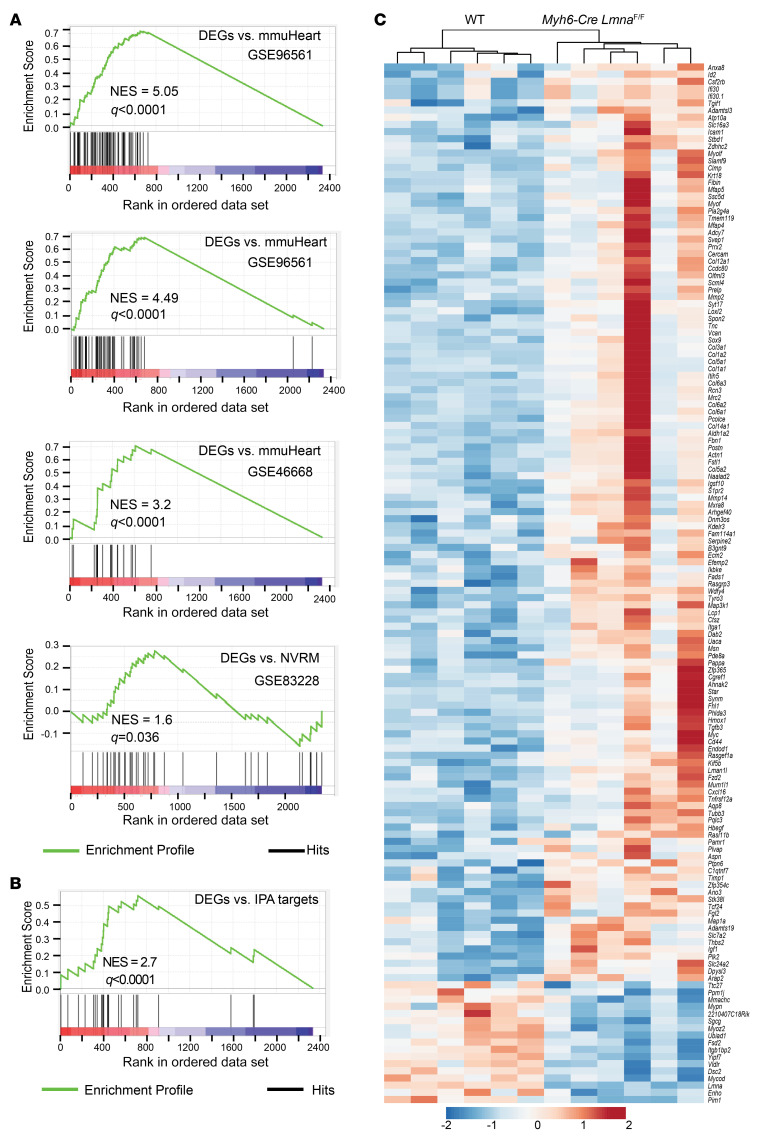
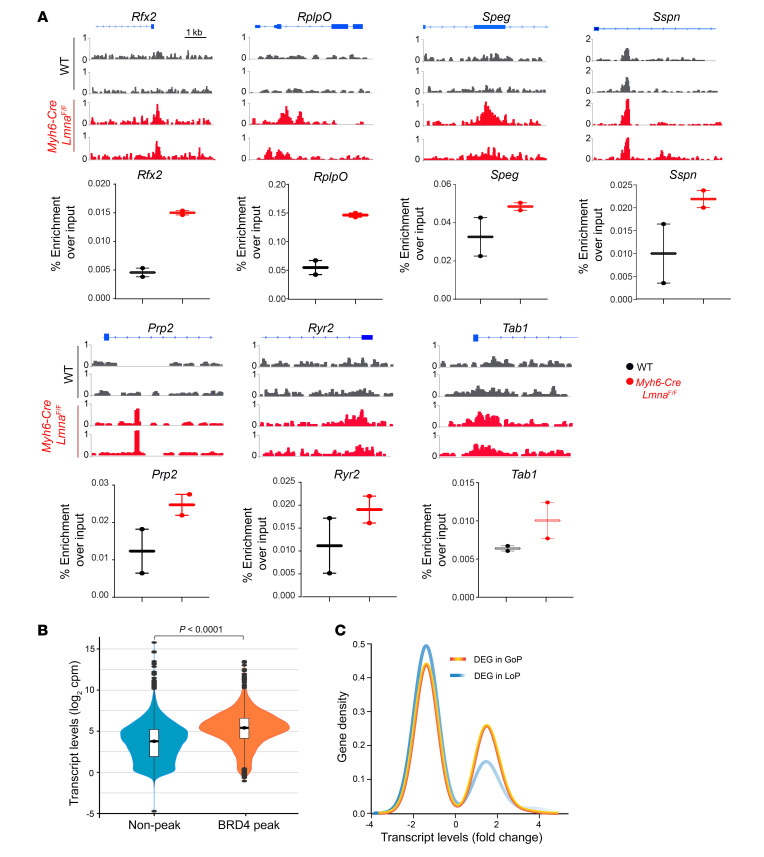
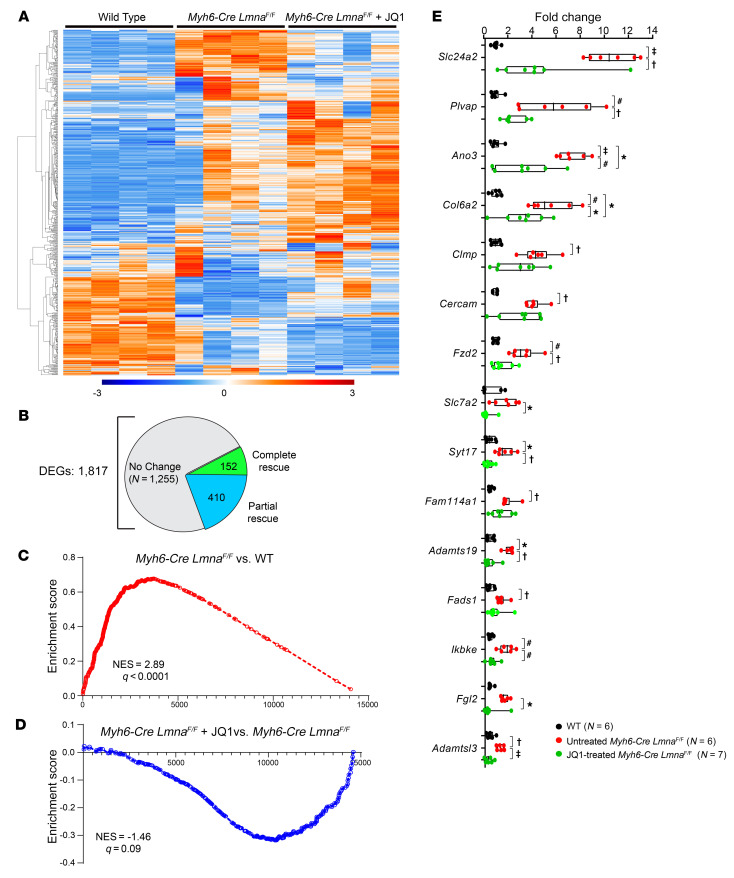
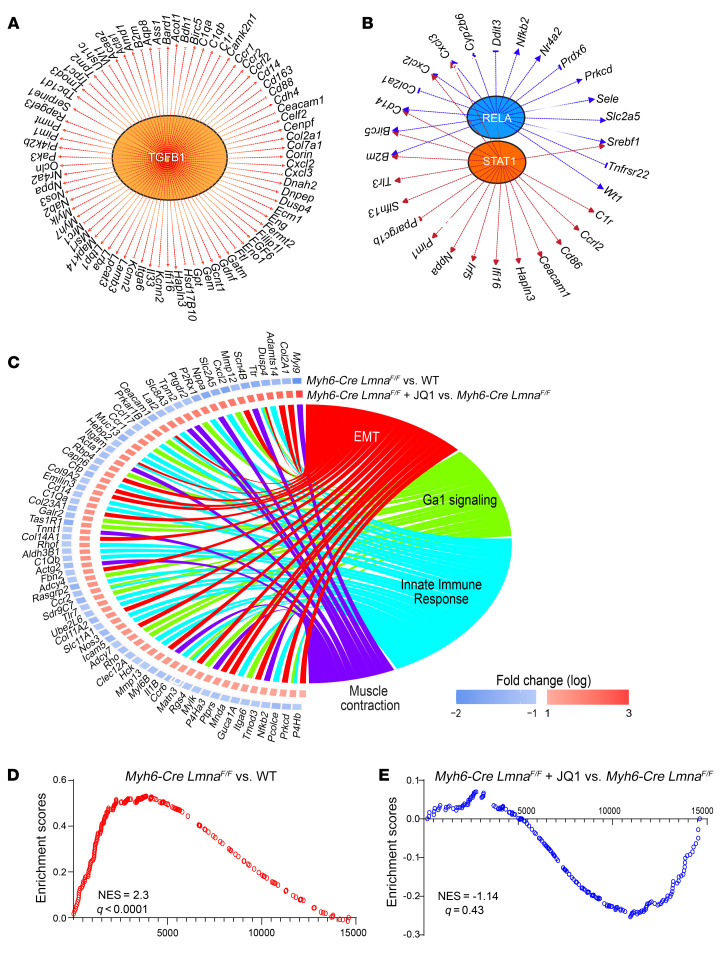
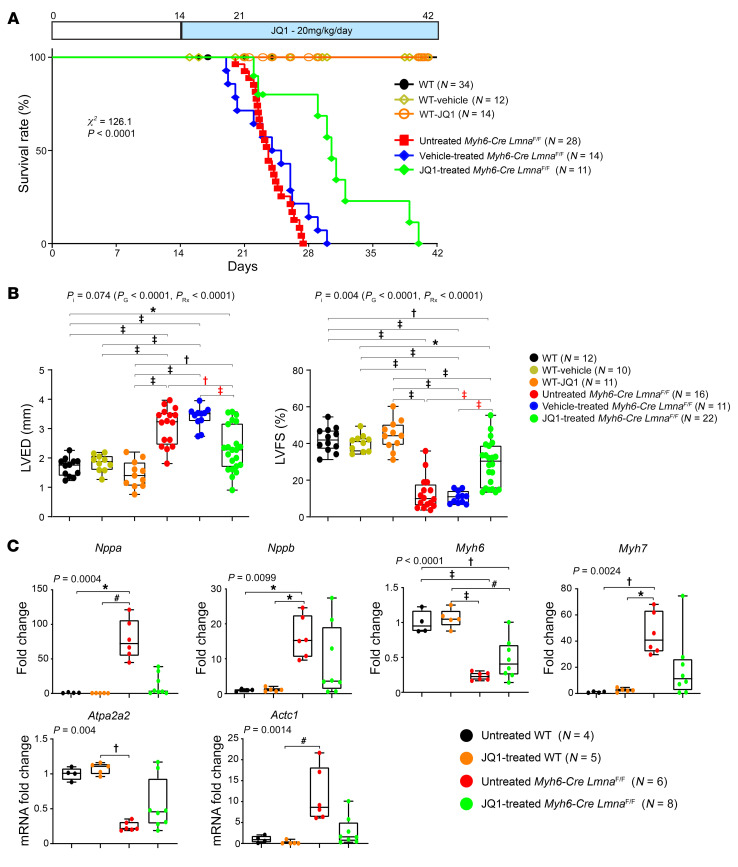
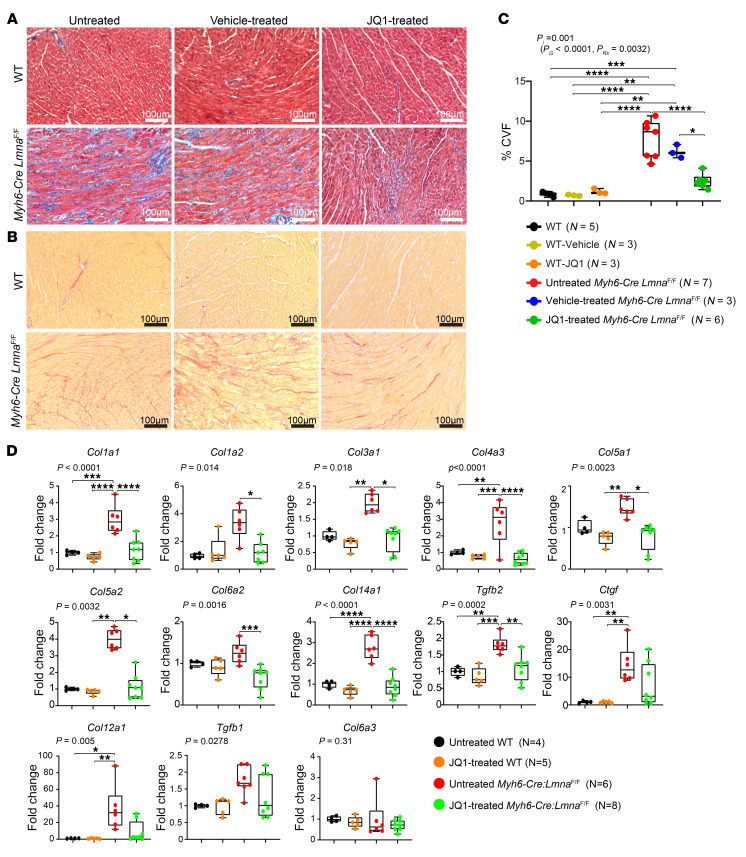
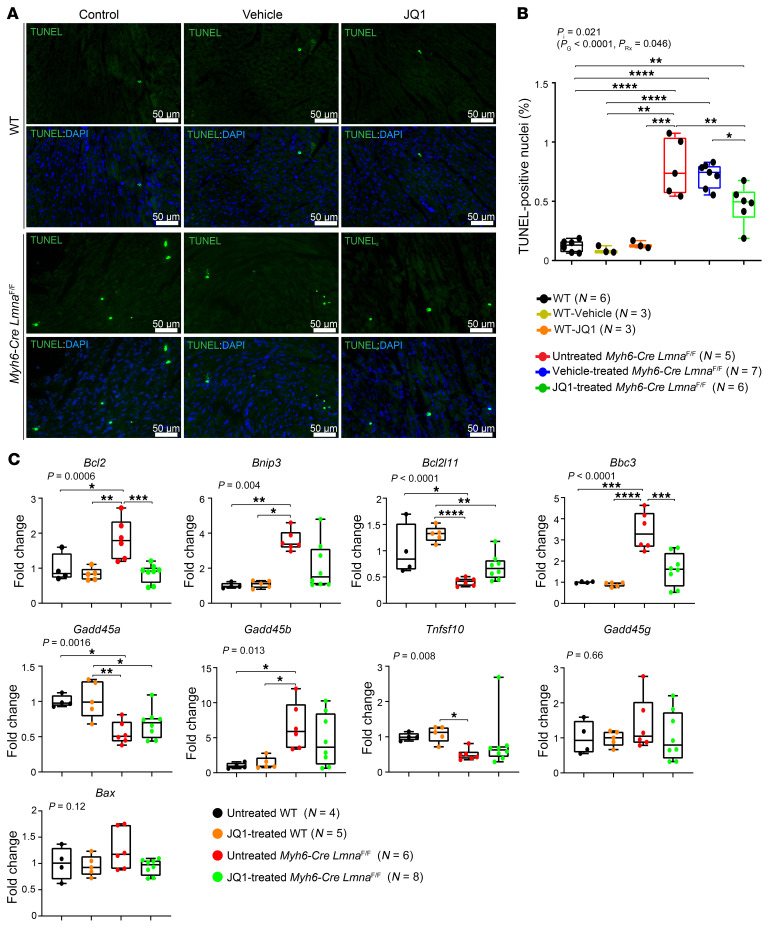
Mutation in the LMNA gene, encoding lamin A/C, causes a diverse group of diseases called laminopathies. Cardiac involvement is the major cause of death and manifests as dilated cardiomyopathy, heart failure, arrhythmias, and sudden death. There is no specific therapy for LMNA-associated cardiomyopathy. We report that deletion of Lmna in cardiomyocytes in mice leads to severe cardiac dysfunction, conduction defect, ventricular arrhythmias, fibrosis, apoptosis, and premature death within 4 weeks. The phenotype is similar to LMNA-associated cardiomyopathy in humans. RNA sequencing, performed before the onset of cardiac dysfunction, led to identification of 2338 differentially expressed genes (DEGs) in Lmna-deleted cardiomyocytes. DEGs predicted activation of bromodomain-containing protein 4 (BRD4), a regulator of chromatin-associated proteins and transcription factors, which was confirmed by complementary approaches, including chromatin immunoprecipitation sequencing. Daily injection of JQ1, a specific BET bromodomain inhibitor, partially reversed the DEGs, including those encoding secretome; improved cardiac function; abrogated cardiac arrhythmias, fibrosis, and apoptosis; and prolonged the median survival time 2-fold in the myocyte-specific Lmna-deleted mice. The findings highlight the important role of LMNA in cardiomyocytes and identify BET bromodomain inhibition as a potential therapeutic target in LMNA-associated cardiomyopathy, for which there is no specific effective therapy.
Keywords: Cardiology; Cardiovascular disease; Fibrosis; Heart failure.
Conflict of interest statement
Figures
Comment in
-
BETs that cover the spread from acquired to heritable heart failure.J Clin Invest. 2020 Sep 1;130(9):4536-4539. doi: 10.1172/JCI140304. J Clin Invest. 2020. PMID: 32773407 Free PMC article.
References
-
- Benjamin EJ, et al. Heart disease and stroke statistics — 2019 update: a report from the American Heart Association. Circulation. 2019;139(10):e56–e528. - PubMed
-
- Metra M, et al. Advanced chronic heart failure: a position statement from the Study Group on Advanced Heart Failure of the Heart Failure Association of the European Society of Cardiology. Eur J Heart Fail. 2007;9(6–7):684–694. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Miscellaneous
