

Inference of Bacterial Small RNA Regulatory Networks and Integration with Transcription Factor-Driven Regulatory Networks
- PMID: 32487739
- PMCID: PMC8534726
- DOI: 10.1128/mSystems.00057-20
Inference of Bacterial Small RNA Regulatory Networks and Integration with Transcription Factor-Driven Regulatory Networks
Abstract
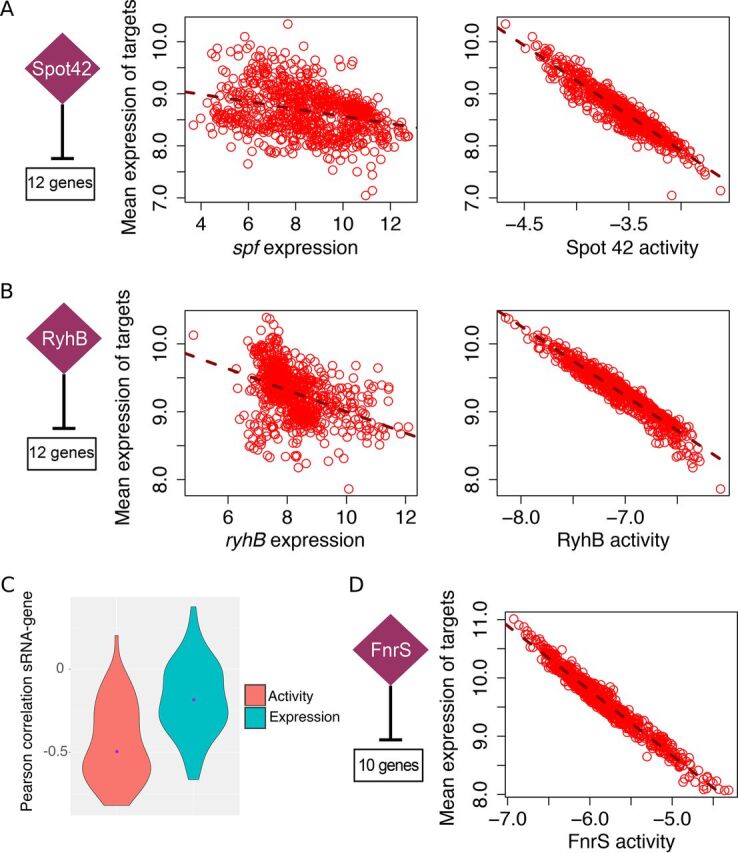
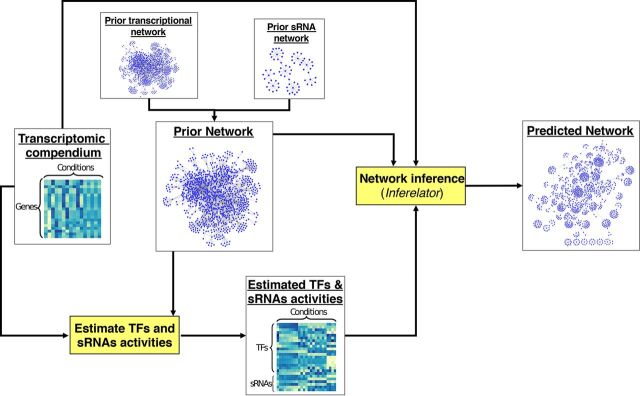
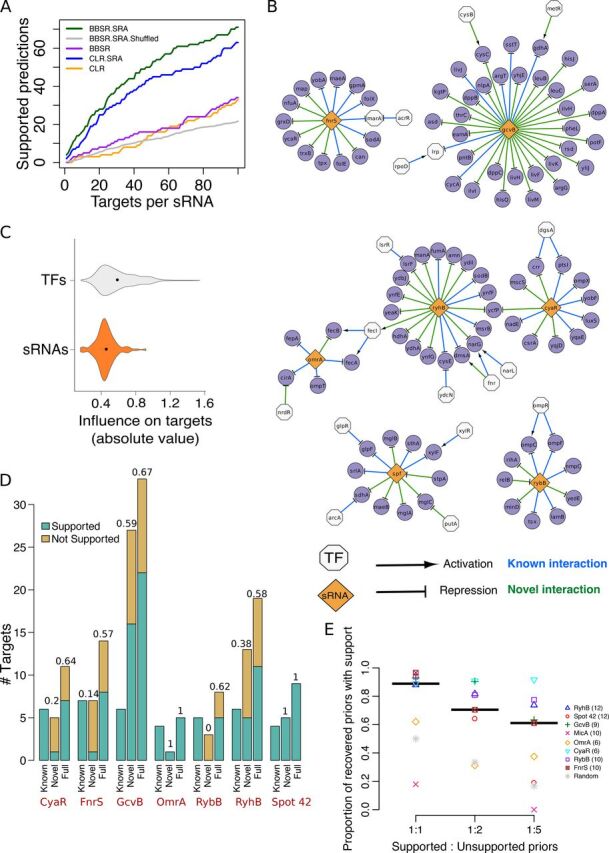
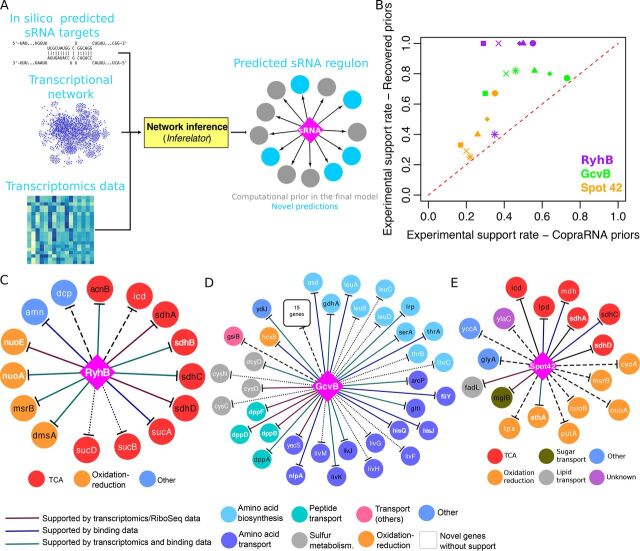
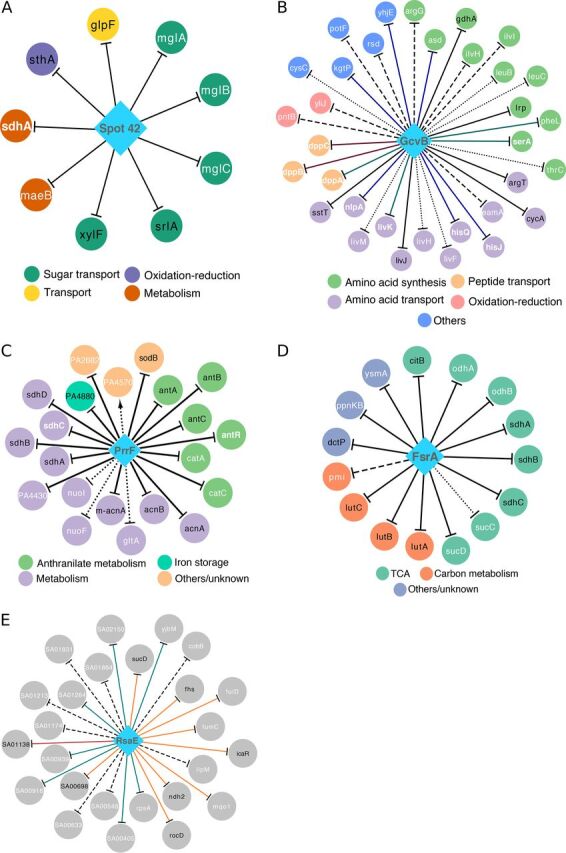
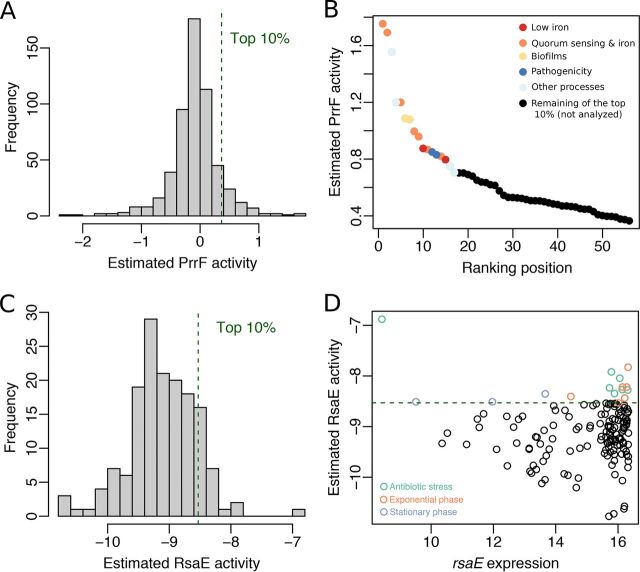
Small noncoding RNAs (sRNAs) are key regulators of bacterial gene expression. Through complementary base pairing, sRNAs affect mRNA stability and translation efficiency. Here, we describe a network inference approach designed to identify sRNA-mediated regulation of transcript levels. We use existing transcriptional data sets and prior knowledge to infer sRNA regulons using our network inference tool, the Inferelator This approach produces genome-wide gene regulatory networks that include contributions by both transcription factors and sRNAs. We show the benefits of estimating and incorporating sRNA activities into network inference pipelines using available experimental data. We also demonstrate how these estimated sRNA regulatory activities can be mined to identify the experimental conditions where sRNAs are most active. We uncover 45 novel experimentally supported sRNA-mRNA interactions in Escherichia coli, outperforming previous network-based efforts. Additionally, our pipeline complements sequence-based sRNA-mRNA interaction prediction methods by adding a data-driven filtering step. Finally, we show the general applicability of our approach by identifying 24 novel, experimentally supported, sRNA-mRNA interactions in Pseudomonas aeruginosa, Staphylococcus aureus, and Bacillus subtilis Overall, our strategy generates novel insights into the functional context of sRNA regulation in multiple bacterial species.IMPORTANCE Individual bacterial genomes can have dozens of small noncoding RNAs with largely unexplored regulatory functions. Although bacterial sRNAs influence a wide range of biological processes, including antibiotic resistance and pathogenicity, our current understanding of sRNA-mediated regulation is far from complete. Most of the available information is restricted to a few well-studied bacterial species; and even in those species, only partial sets of sRNA targets have been characterized in detail. To close this information gap, we developed a computational strategy that takes advantage of available transcriptional data and knowledge about validated and putative sRNA-mRNA interactions for inferring expanded sRNA regulons. Our approach facilitates the identification of experimentally supported novel interactions while filtering out false-positive results. Due to its data-driven nature, our method prioritizes biologically relevant interactions among lists of candidate sRNA-target pairs predicted in silico from sequence analysis or derived from sRNA-mRNA binding experiments.
Keywords: gene networks; global regulation; small RNAs.
Copyright © 2020 Arrieta-Ortiz et al.
Figures
References
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Miscellaneous
