

Loss of Protease-Activated Receptor 4 Prevents Inflammation Resolution and Predisposes the Heart to Cardiac Rupture After Myocardial Infarction
- PMID: 32489148
- PMCID: PMC9341277
- DOI: 10.1161/CIRCULATIONAHA.119.044340
Loss of Protease-Activated Receptor 4 Prevents Inflammation Resolution and Predisposes the Heart to Cardiac Rupture After Myocardial Infarction
Abstract
Background: Cardiac rupture is a major lethal complication of acute myocardial infarction (MI). Despite significant advances in reperfusion strategies, mortality from cardiac rupture remains high. Studies suggest that cardiac rupture can be accelerated by thrombolytic therapy, but the relevance of this risk factor remains controversial.
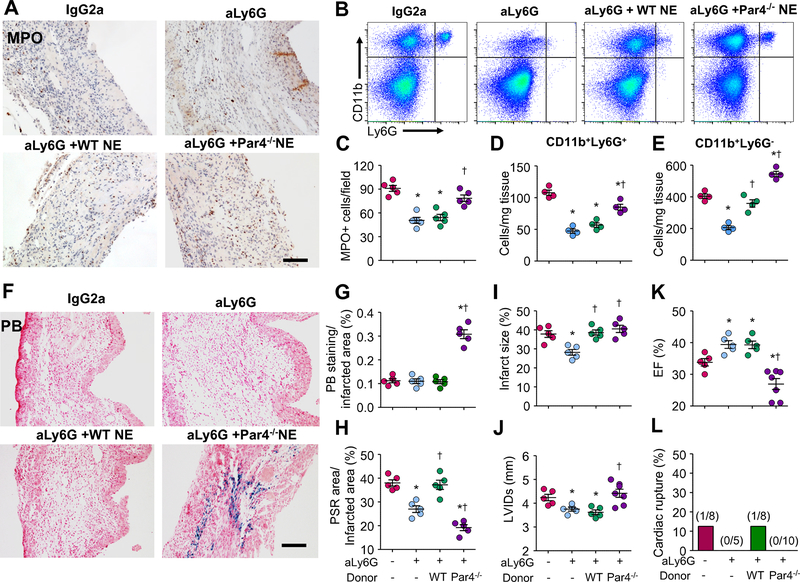
Methods: We analyzed protease-activated receptor 4 (Par4) expression in mouse hearts with MI and investigated the effects of Par4 deletion on cardiac remodeling and function after MI by echocardiography, quantitative immunohistochemistry, and flow cytometry.
Results: Par4 mRNA and protein levels were increased in mouse hearts after MI and in isolated cardiomyocytes in response to hypertrophic and inflammatory stimuli. Par4-deficient mice showed less myocyte apoptosis, reduced infarct size, and improved functional recovery after acute MI relative to wild-type (WT). Conversely, Par4-/- mice showed impaired cardiac function, greater rates of myocardial rupture, and increased mortality after chronic MI relative to WT. Pathological evaluation of hearts from Par4-/- mice demonstrated a greater infarct expansion, increased cardiac hemorrhage, and delayed neutrophil accumulation, which resulted in impaired post-MI healing compared with WT. Par4 deficiency also attenuated neutrophil apoptosis in vitro and after MI in vivo and impaired inflammation resolution in infarcted myocardium. Transfer of Par4-/- neutrophils, but not of Par4-/- platelets, in WT recipient mice delayed inflammation resolution, increased cardiac hemorrhage, and enhanced cardiac dysfunction. In parallel, adoptive transfer of WT neutrophils into Par4-/- mice restored inflammation resolution, reduced cardiac rupture incidence, and improved cardiac function after MI.
Conclusions: These findings reveal essential roles of Par4 in neutrophil apoptosis and inflammation resolution during myocardial healing and point to Par4 inhibition as a potential therapy that should be limited to the acute phases of ischemic insult and avoided for long-term treatment after MI.
Keywords: heart rupture; inflammation; myocardial infarction; neutrophils; protease-activated receptor 4.
Figures







References
-
- Institute NHLaB. Morbidity and mortality: 2012 chart book on cardiovascular and lung diseases. Bethesda, MD: National Institute of Health. 2012
-
- Becker RC, Gore JM, Lambrew C, Douglas Weaver W, Michael Rubison R, French WJ, Tiefenbrunn AJ, Bowlby LJ, Rogers WJ. A composite view of cardiac rupture in the united states national registry of myocardial infarction. J Am Coll Cardiol. 1996;27:1321–1326 - PubMed
-
- Gao X-M, White DA, Dart AM, Du X-J. Post-infarct cardiac rupture: Recent insights on pathogenesis and therapeutic interventions. Pharmacol Therap. 2012;134:156–179 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
