

A mouse SWATH-mass spectrometry reference spectral library enables deconvolution of species-specific proteomic alterations in human tumour xenografts
- PMID: 32493768
- PMCID: PMC7375474
- DOI: 10.1242/dmm.044586
A mouse SWATH-mass spectrometry reference spectral library enables deconvolution of species-specific proteomic alterations in human tumour xenografts
Abstract
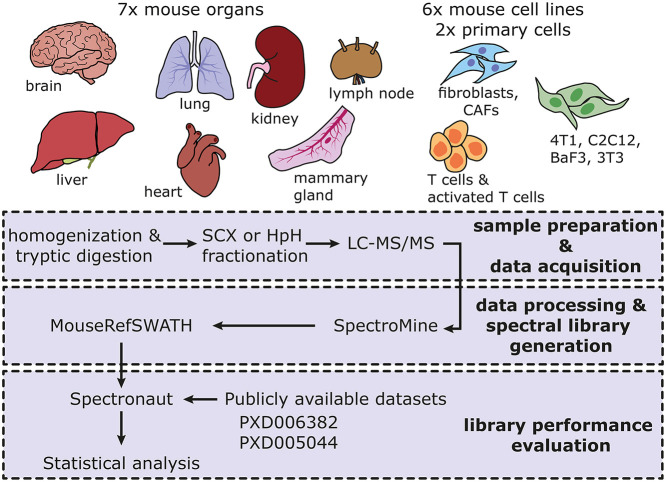
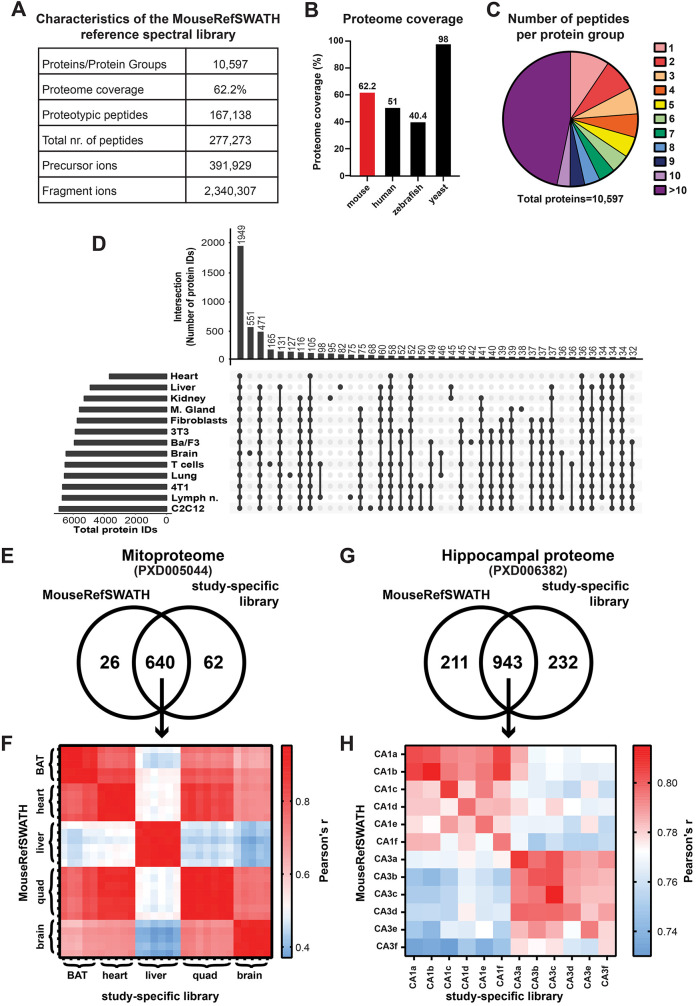
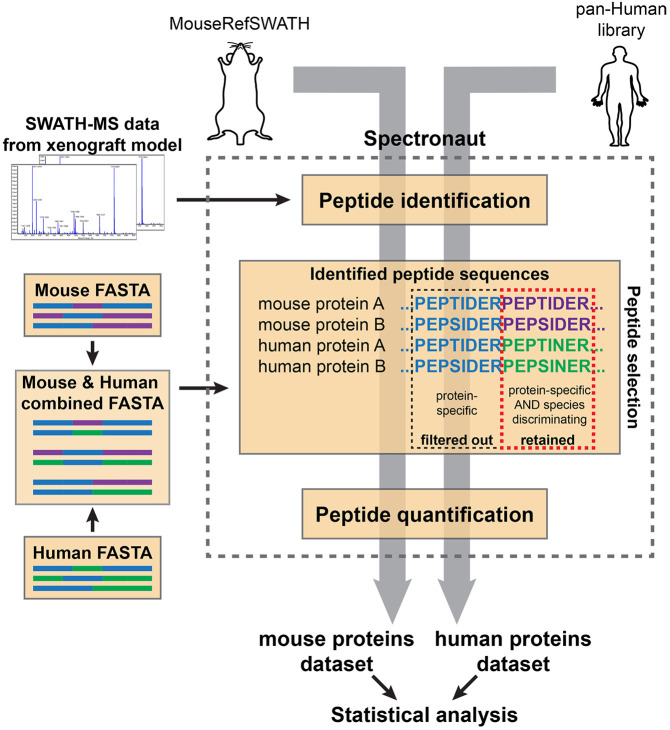
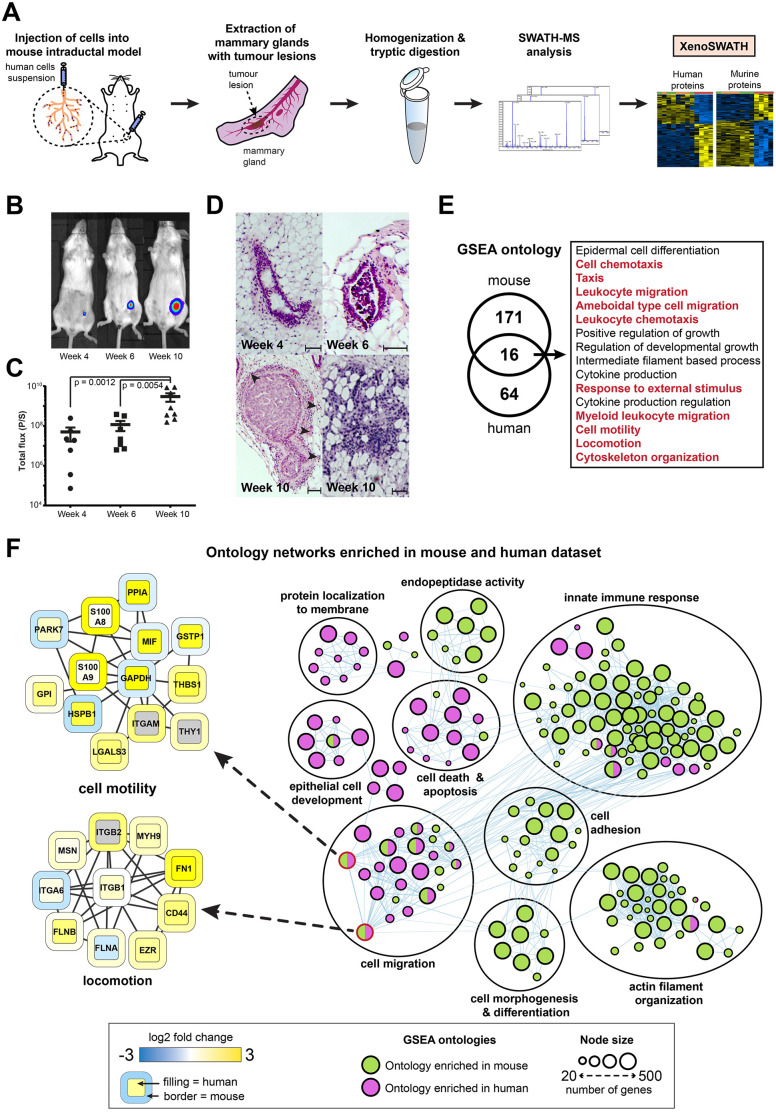
SWATH-mass spectrometry (MS) enables accurate and reproducible proteomic profiling in multiple model organisms including the mouse. Here, we present a comprehensive mouse reference spectral library (MouseRefSWATH) that permits quantification of up to 10,597 proteins (62.2% of the mouse proteome) by SWATH-MS. We exploit MouseRefSWATH to develop an analytical pipeline for species-specific deconvolution of proteomic alterations in human tumour xenografts (XenoSWATH). This method overcomes the challenge of high sequence similarity between mouse and human proteins, facilitating the study of host microenvironment-tumour interactions from 'bulk tumour' measurements. We apply the XenoSWATH pipeline to characterize an intraductal xenograft model of breast ductal carcinoma in situ and uncover complex regulation consistent with stromal reprogramming, where the modulation of cell migration pathways is not restricted to tumour cells but also operates in the mouse stroma upon progression to invasive disease. MouseRefSWATH and XenoSWATH open new opportunities for in-depth and reproducible proteomic assessment to address wide-ranging biological questions involving this important model organism.
Keywords: Breast cancer; DCIS; Mass spectrometry; Mouse; Proteomics; SWATH-MS; Xenografts.
© 2020. Published by The Company of Biologists Ltd.
Conflict of interest statement
Competing interestsThe authors declare no competing or financial interests.
Figures
References
-
- Barkovits K., Pacharra S., Pfeiffer K., Steinbach S., Eisenacher M., Marcus K. and Uszkoreit J. (2020). Reproducibility, specificity and accuracy of relative quantification using spectral library-based data-independent acquisition. Mol. Cell. Proteomics 19, 181-197. 10.1074/mcp.RA119.001714 - DOI - PMC - PubMed
-
- Behbod F., Kittrell F. S., Lamarca H., Edwards D., Kerbawy S., Heestand J. C., Young E., Mukhopadhyay P., Yeh H. W., Allred D. C. et al. (2009). An intraductal human-in-mouse transplantation model mimics the subtypes of ductal carcinoma in situ. Breast Cancer Res. 11, R66 10.1186/bcr2358 - DOI - PMC - PubMed
-
- Benjamini Y. and Hochberg Y. (1995). Controlling the false discovery rate - a practical and powerful approach to multiple testing. J.R. Stat. Soc. B 57, 289-300. 10.1111/j.2517-6161.1995.tb02031.x - DOI
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
