

Polyamine regulation of ion channel assembly and implications for nicotinic acetylcholine receptor pharmacology
- PMID: 32493979
- PMCID: PMC7271128
- DOI: 10.1038/s41467-020-16629-3
Polyamine regulation of ion channel assembly and implications for nicotinic acetylcholine receptor pharmacology
Abstract
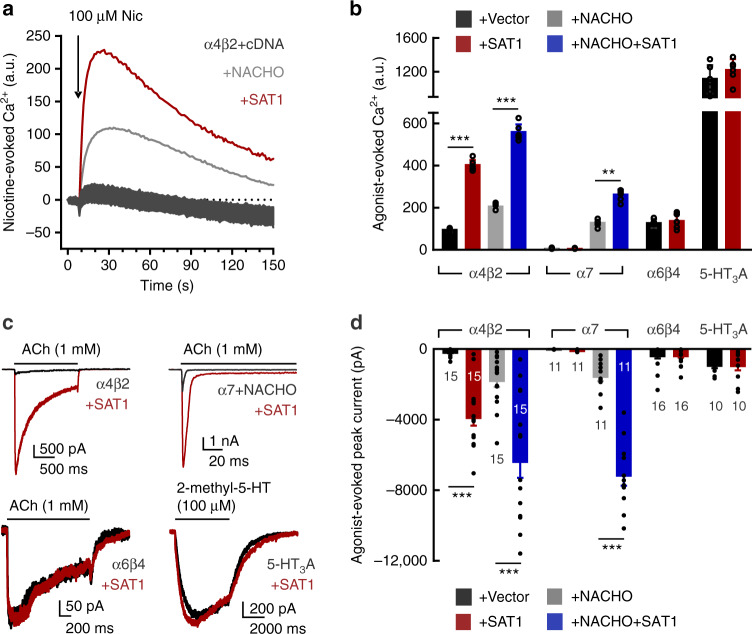
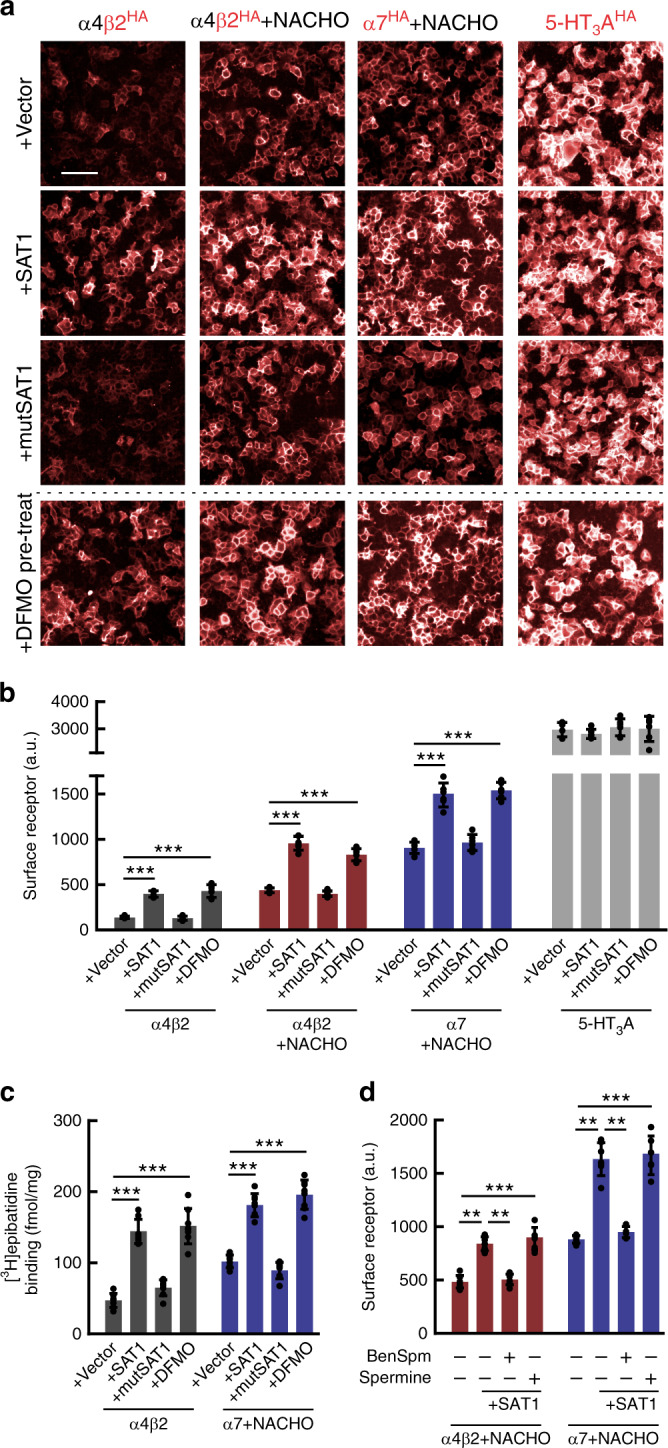
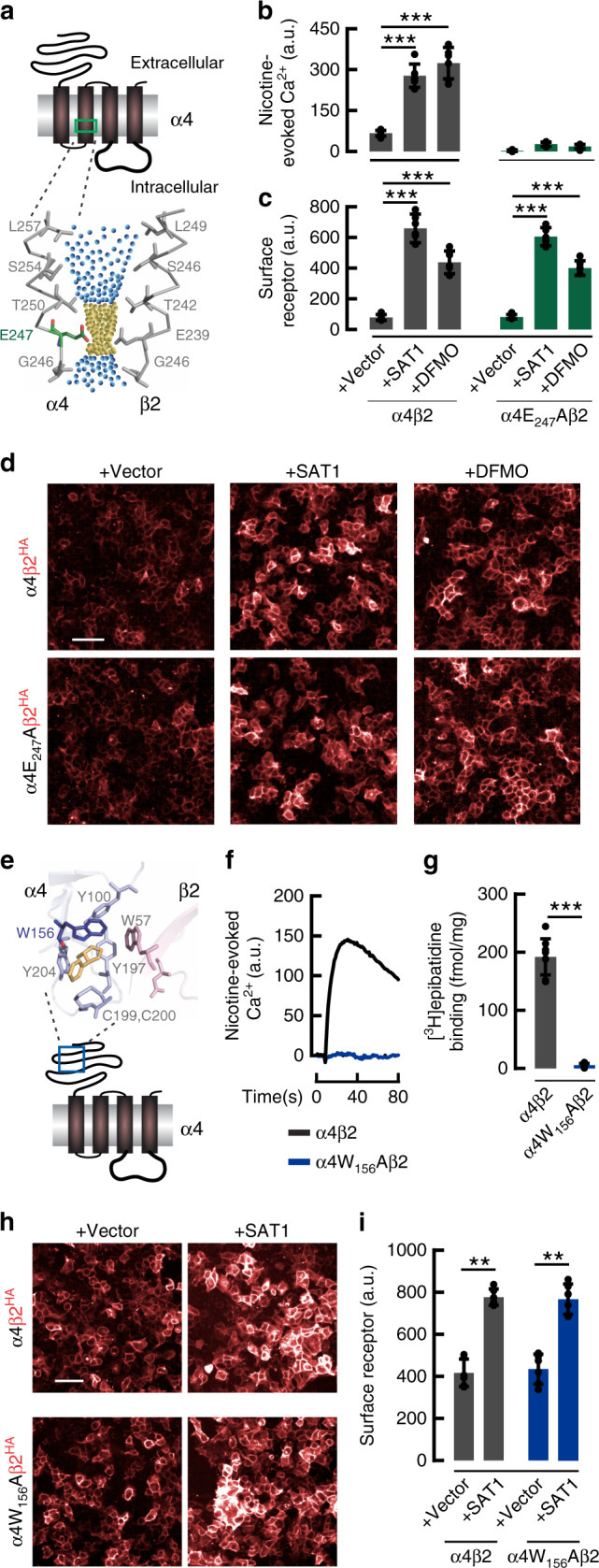
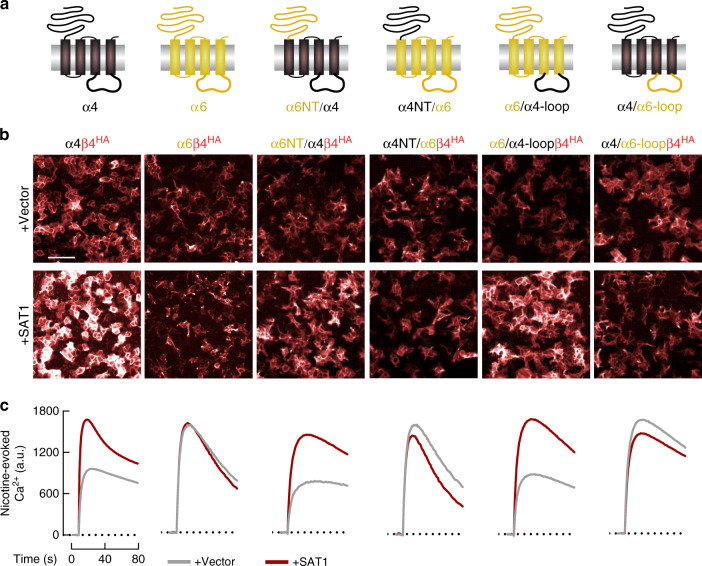
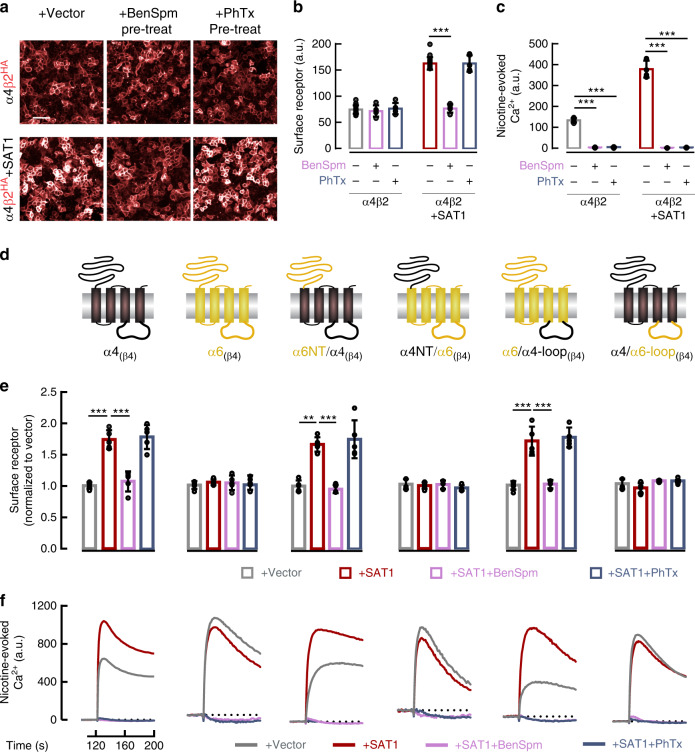
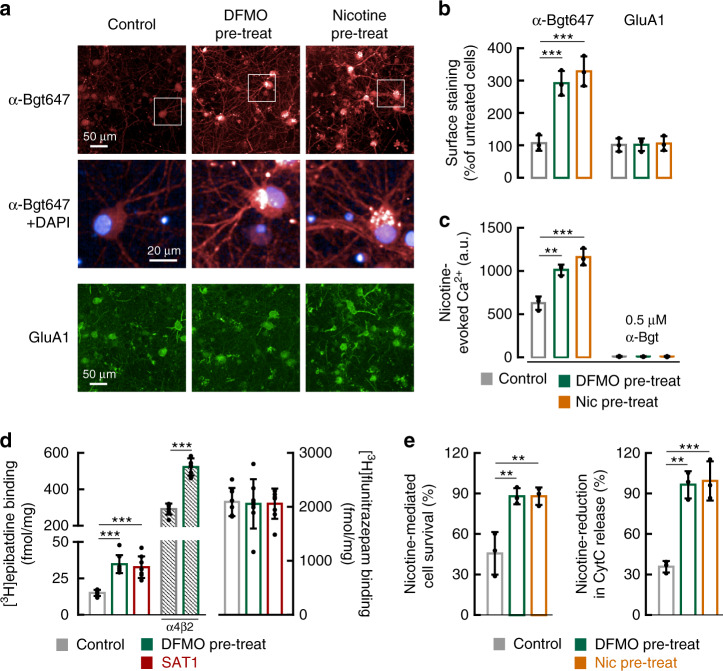
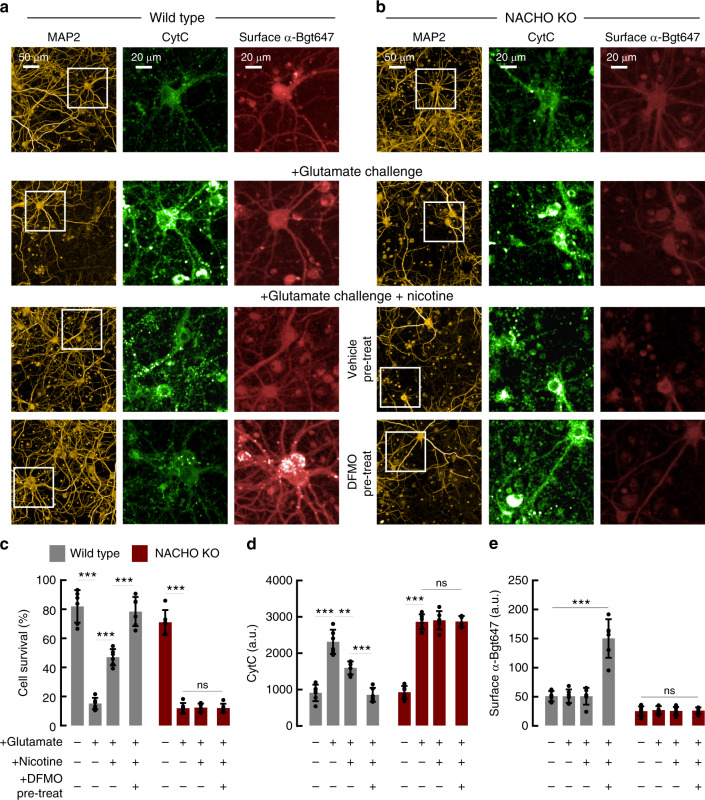
Small molecule polyamines are abundant in all life forms and participate in diverse aspects of cell growth and differentiation. Spermidine/spermine acetyltransferase (SAT1) is the rate-limiting enzyme in polyamine catabolism and a primary genetic risk factor for suicidality. Here, using genome-wide screening, we find that SAT1 selectively controls nicotinic acetylcholine receptor (nAChR) biogenesis. SAT1 specifically augments assembly of nAChRs containing α7 or α4β2, but not α6 subunits. Polyamines are classically studied as regulators of ion channel gating that engage the nAChR channel pore. In contrast, we find polyamine effects on assembly involve the nAChR cytosolic loop. Neurological studies link brain polyamines with neurodegenerative conditions. Our pharmacological and transgenic animal studies find that reducing polyamines enhances cortical neuron nAChR expression and augments nicotine-mediated neuroprotection. Taken together, we describe a most unexpected role for polyamines in regulating ion channel assembly, which provides a new avenue for nAChR neuropharmacology.
Conflict of interest statement
All contributing authors are full-time employees in Johnson and Johnson.
Figures
References
-
- Role LW, Berg DK. Nicotinic receptors in the development and modulation of CNS synapses. Neuron. 1996;16:1077–1085. - PubMed
-
- Gotti C, Clementi F. Neuronal nicotinic receptors: from structure to pathology. Prog. Neurobiol. 2004;74:363–396. - PubMed
-
- Lindstrom J. Nicotinic acetylcholine receptors in health and disease. Mol. Neurobiol. 1997;15:193–222. - PubMed
-
- Hogg RC, Raggenbass M, Bertrand D. Nicotinic acetylcholine receptors: from structure to brain function. Rev. Physiol., Biochem. Pharmacol. 2003;147:1–46. - PubMed
-
- Le Novere N, Corringer PJ, Changeux JP. The diversity of subunit composition in nAChRs: evolutionary origins, physiologic and pharmacologic consequences. J. Neurobiol. 2002;53:447–456. - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
