

The CDK inhibitor CR8 acts as a molecular glue degrader that depletes cyclin K
- PMID: 32494016
- PMCID: PMC7486275
- DOI: 10.1038/s41586-020-2374-x
The CDK inhibitor CR8 acts as a molecular glue degrader that depletes cyclin K
Abstract
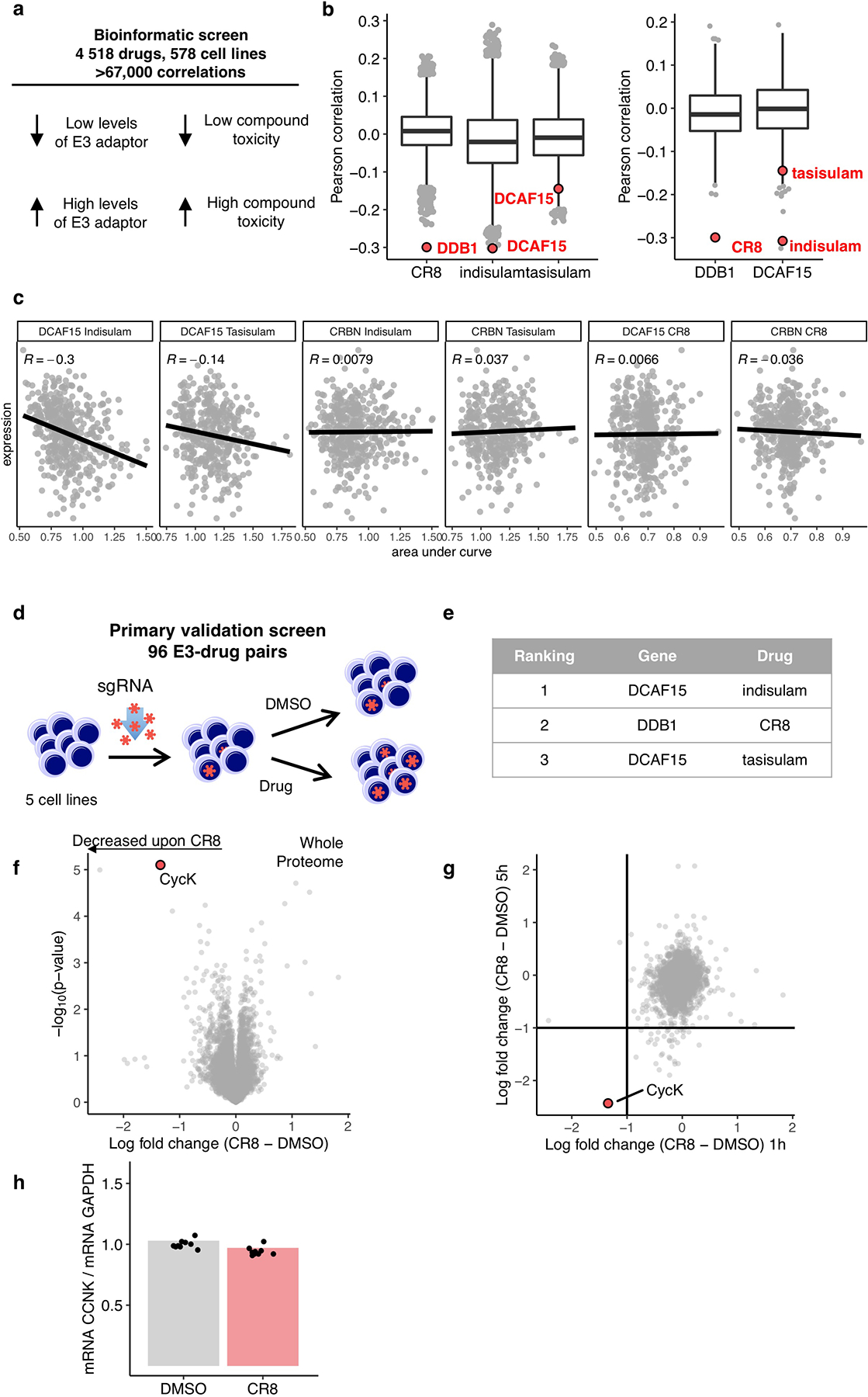
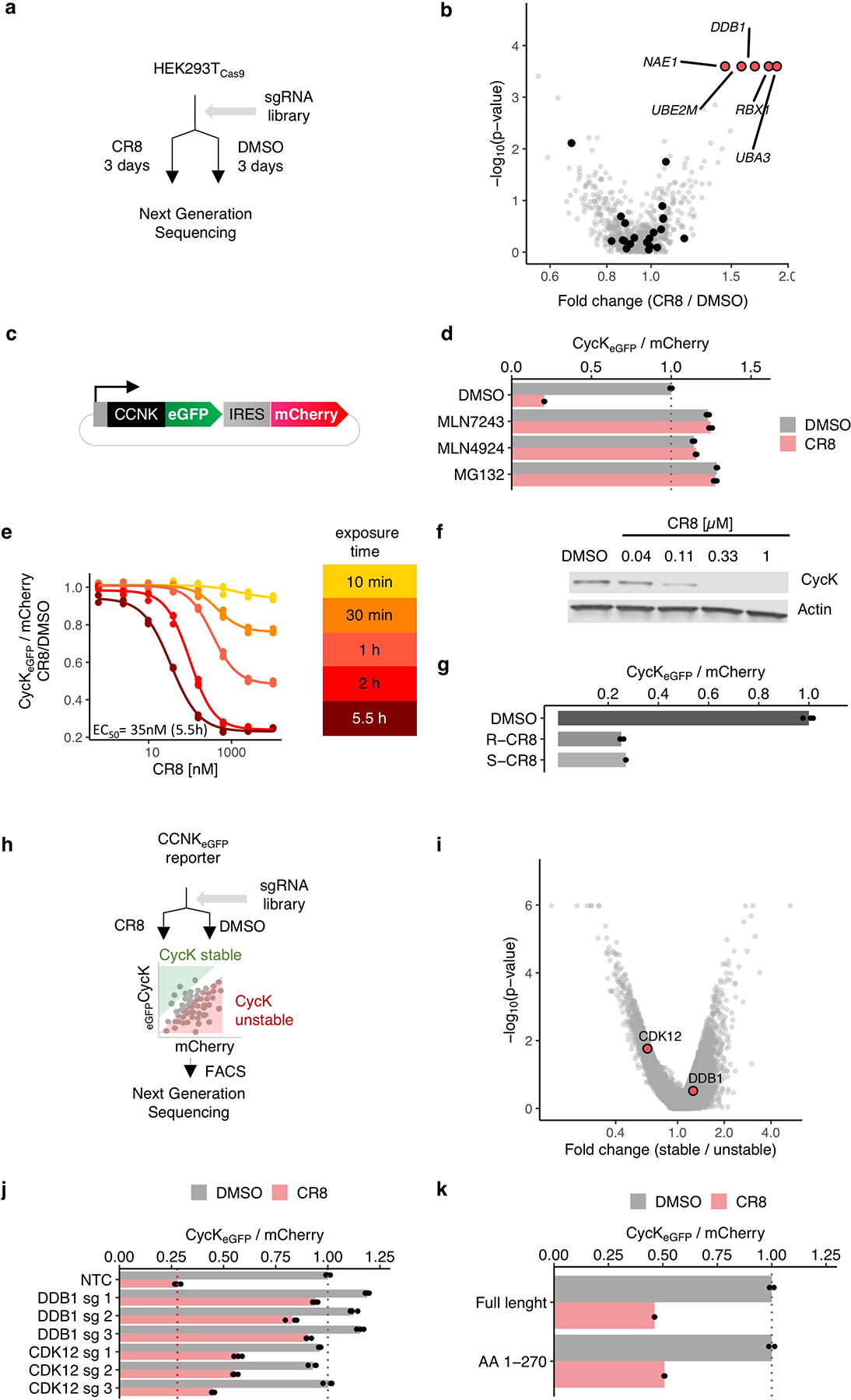
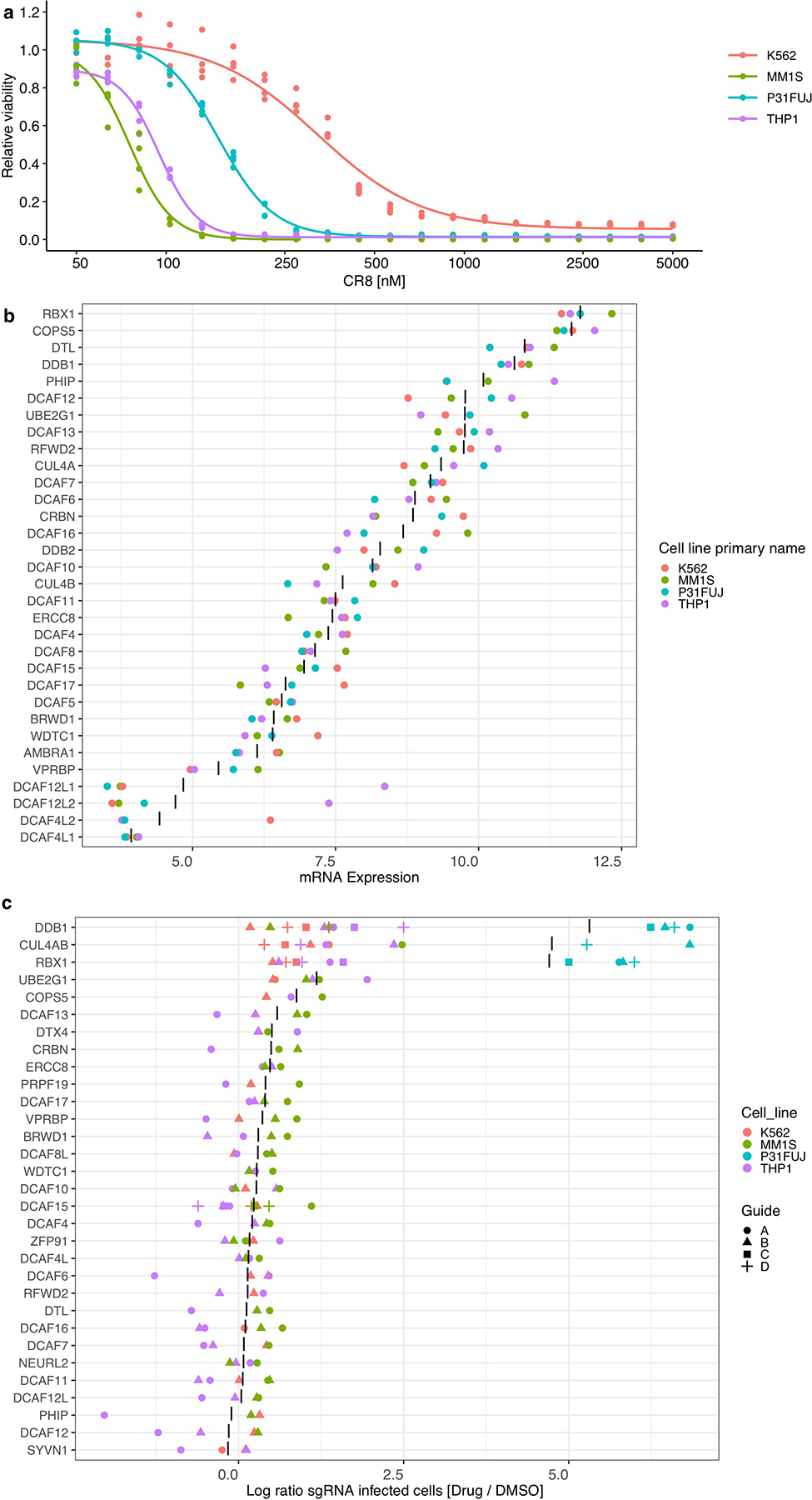
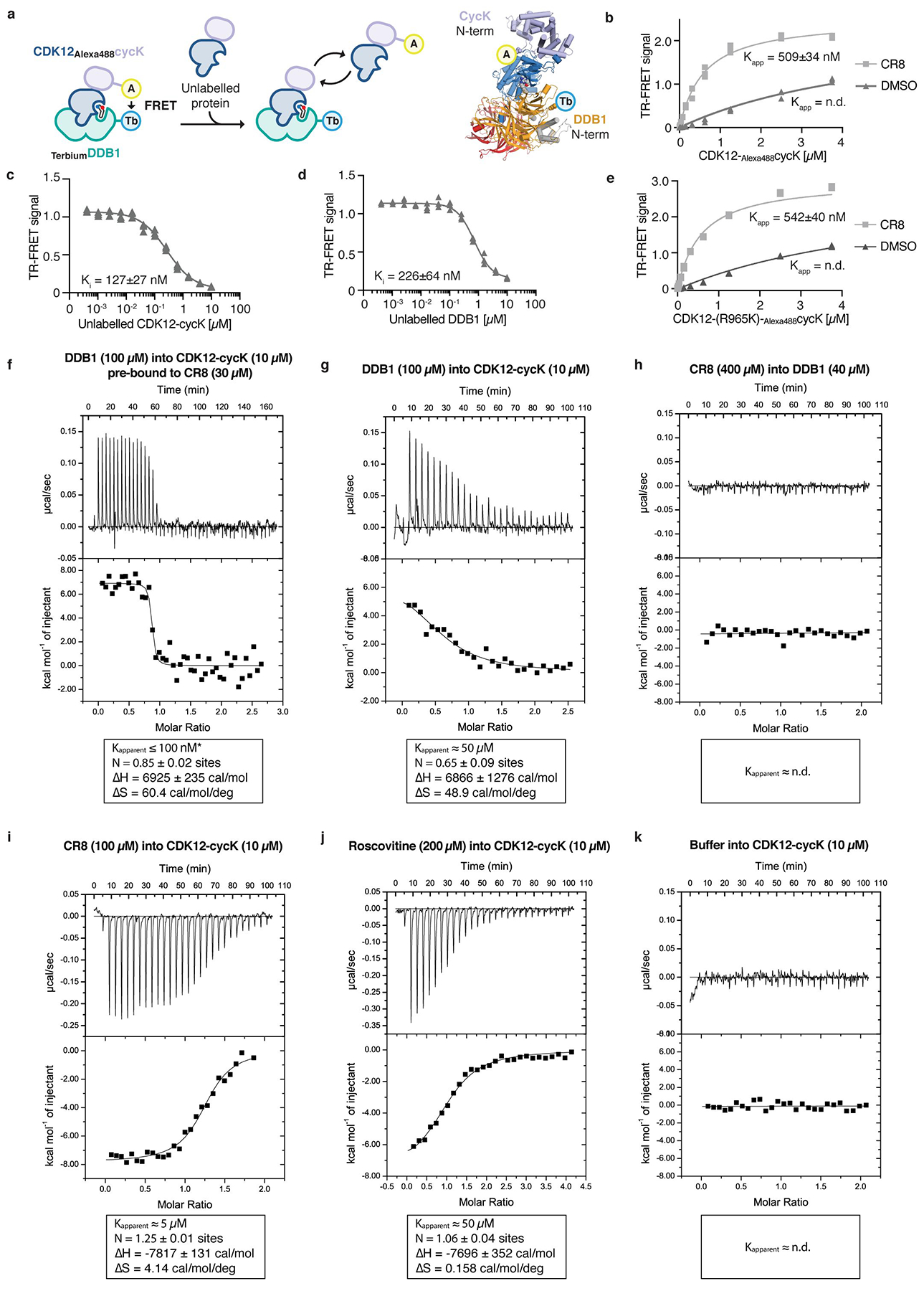
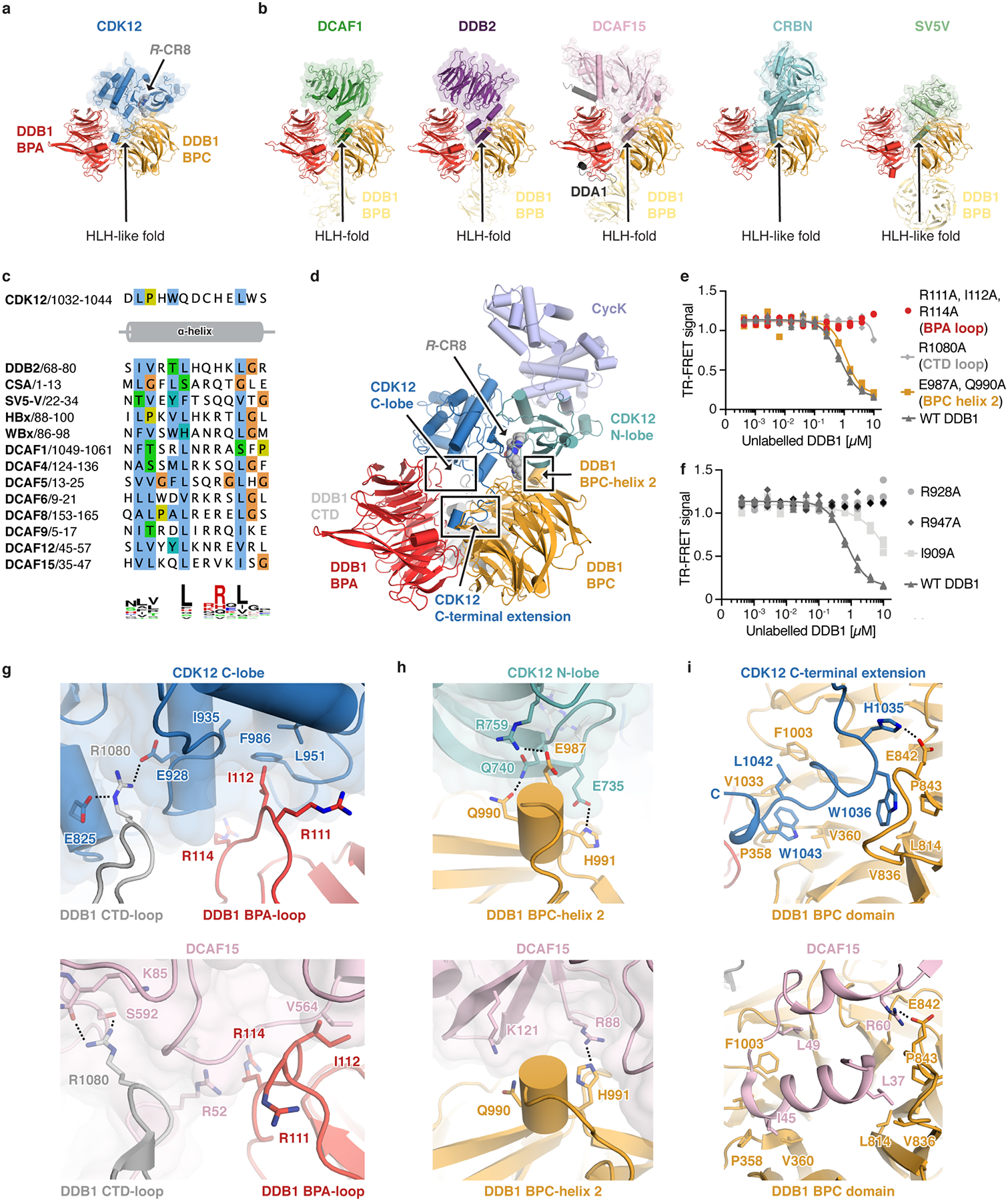
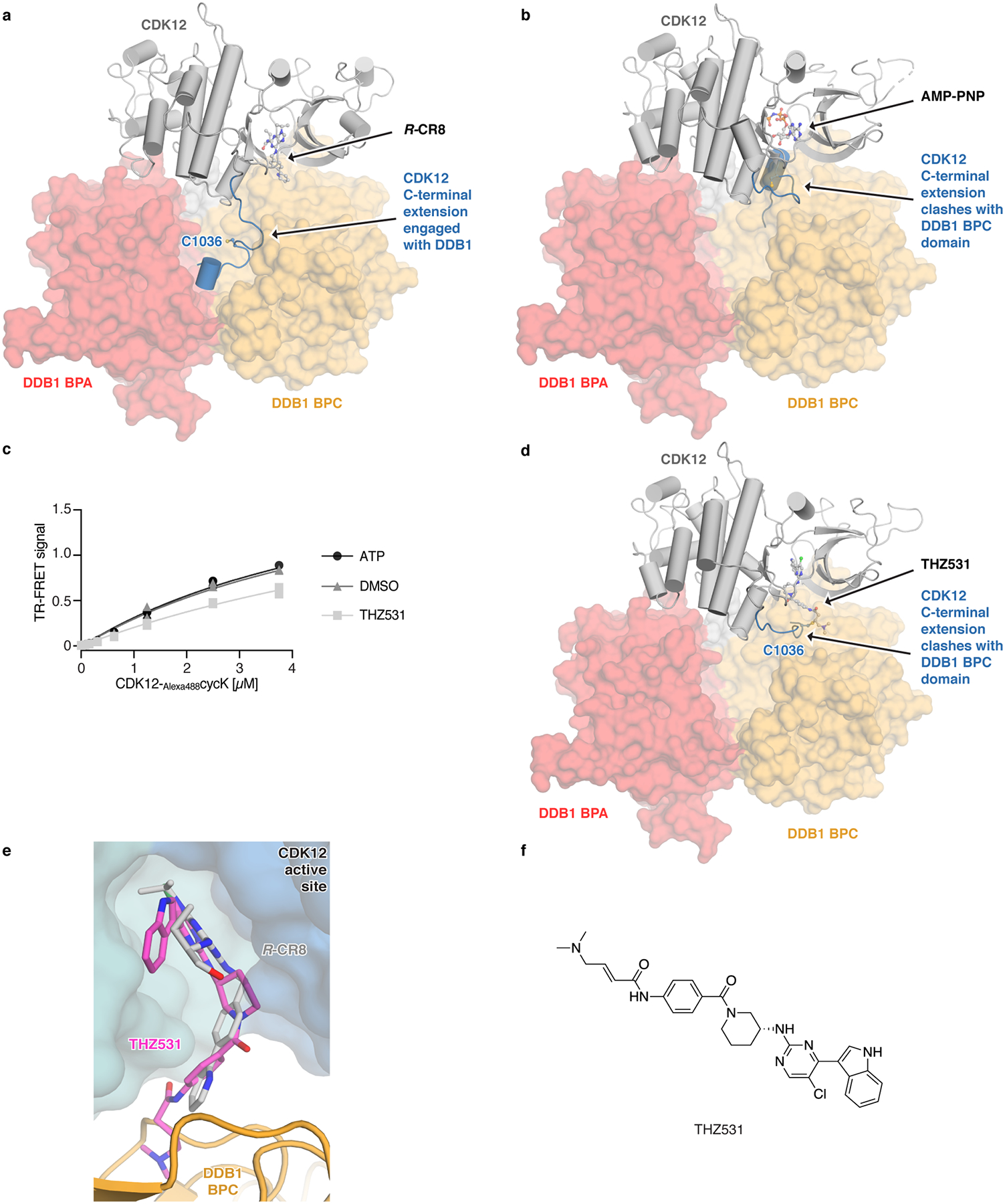
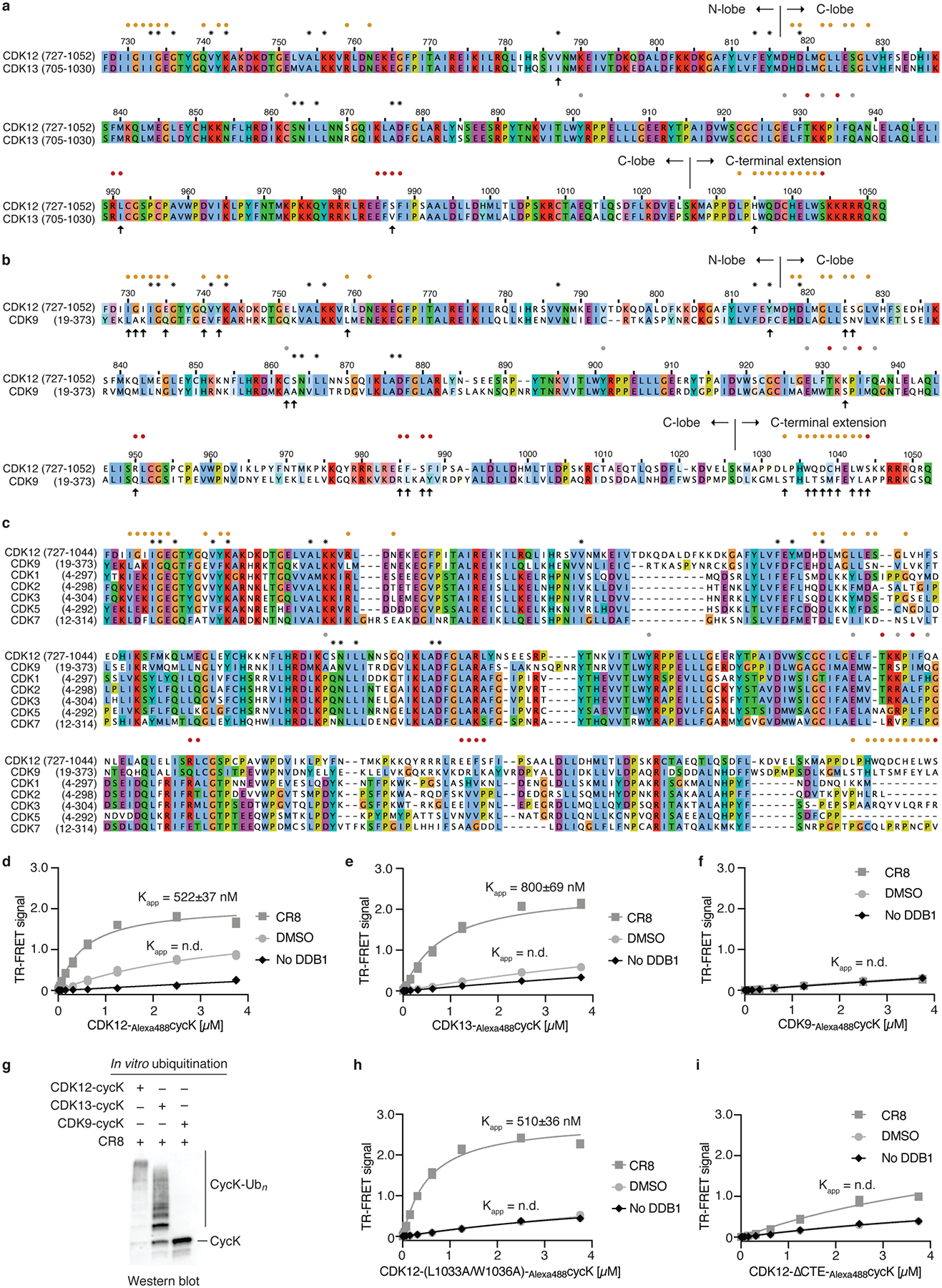
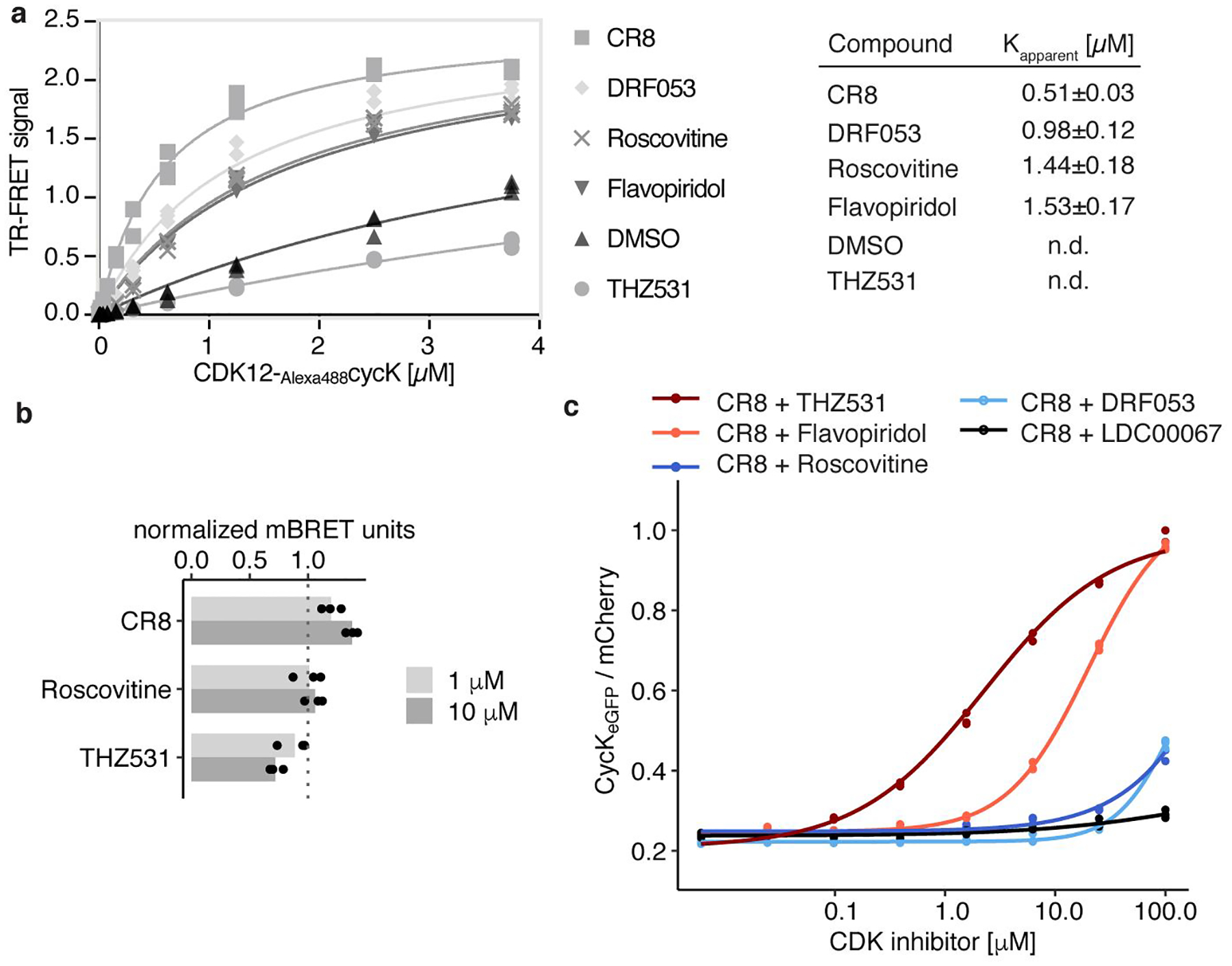
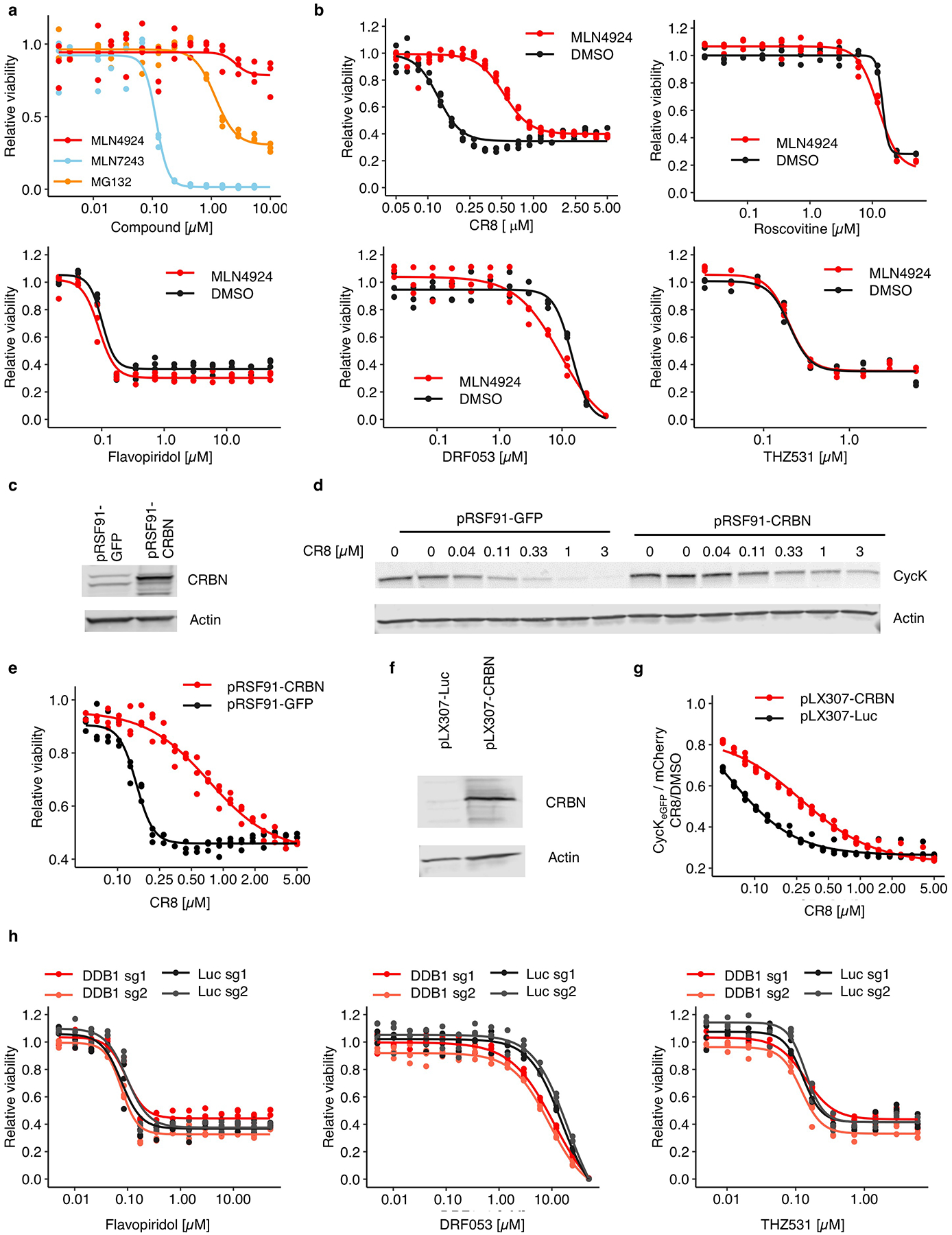
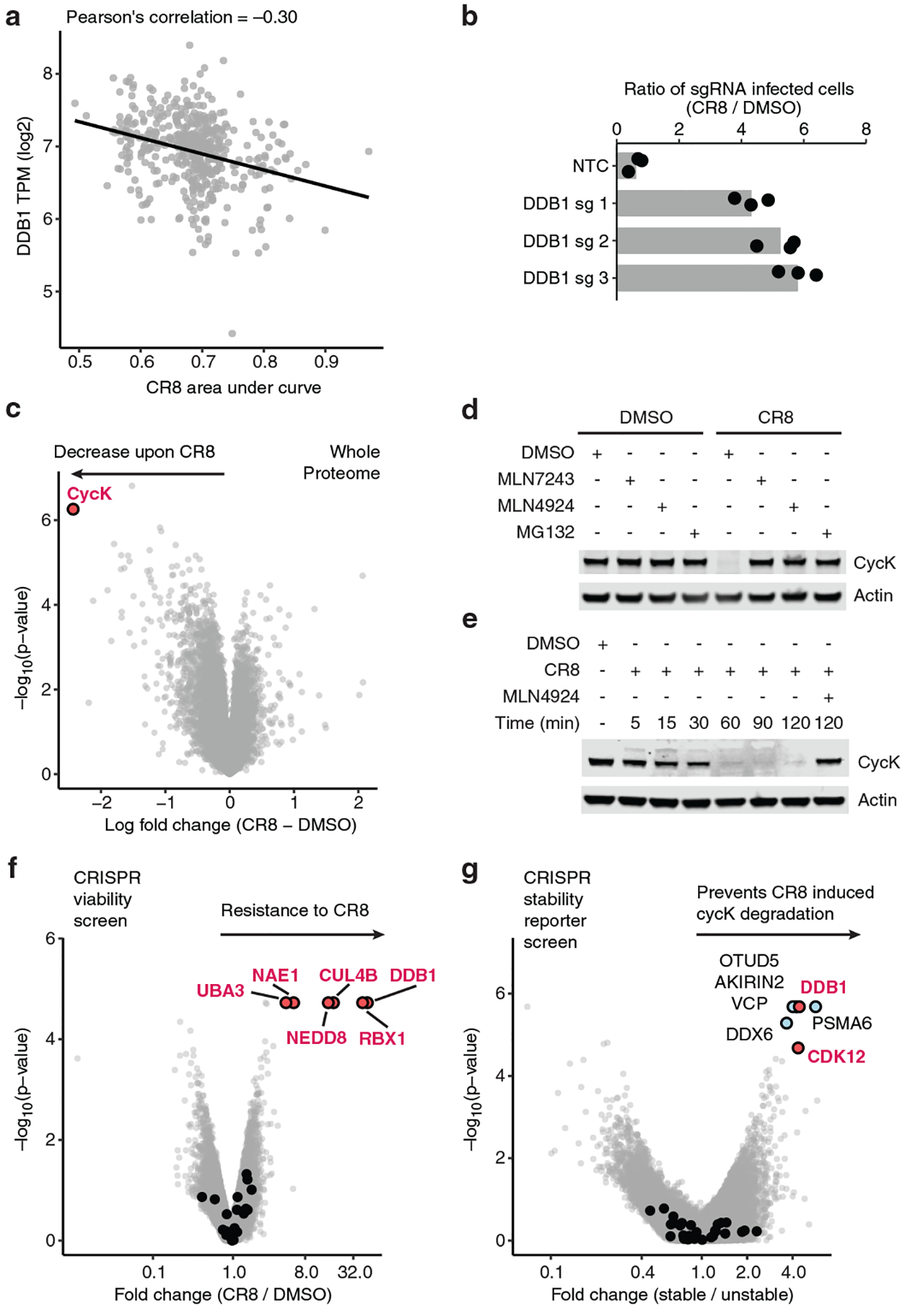
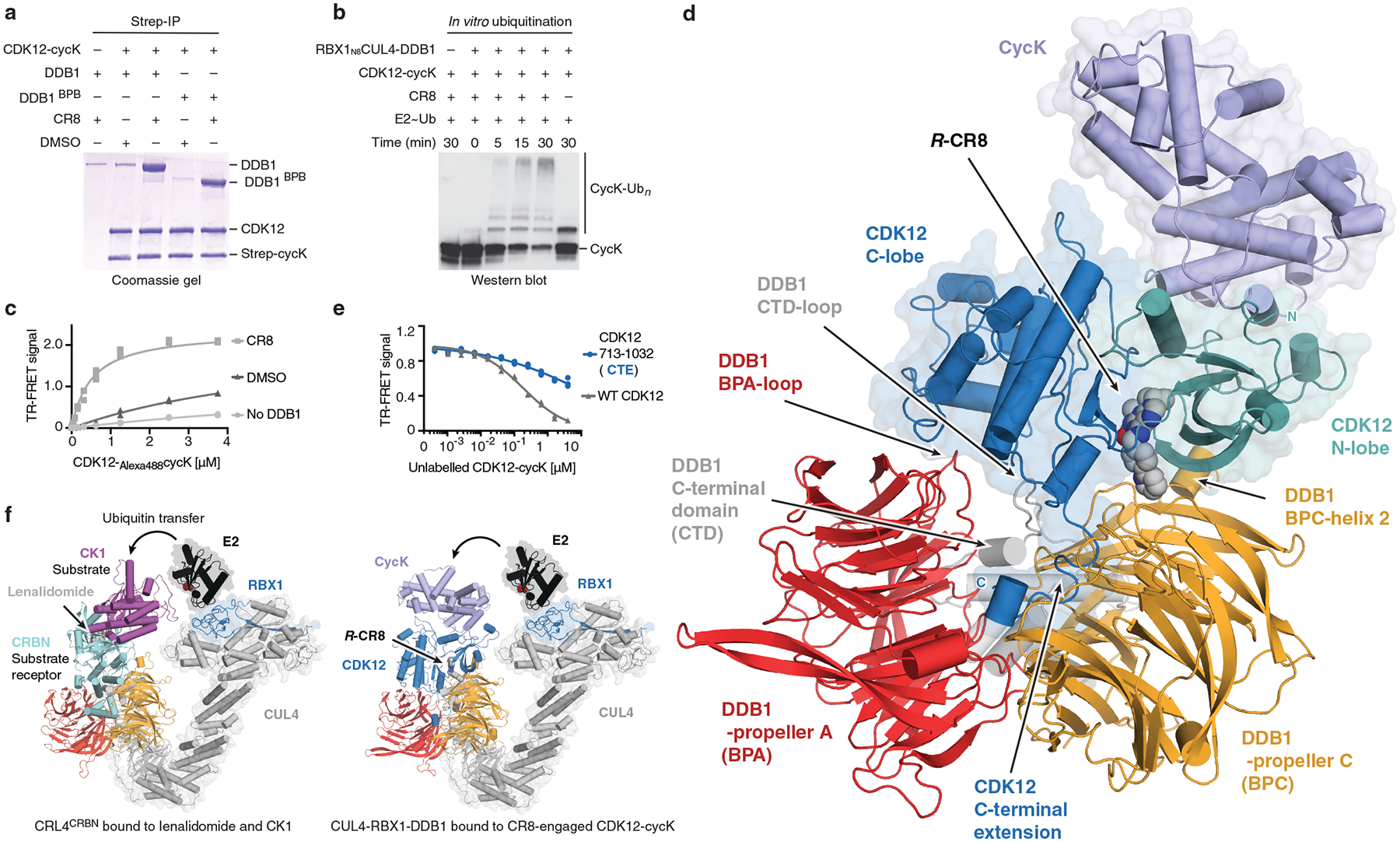
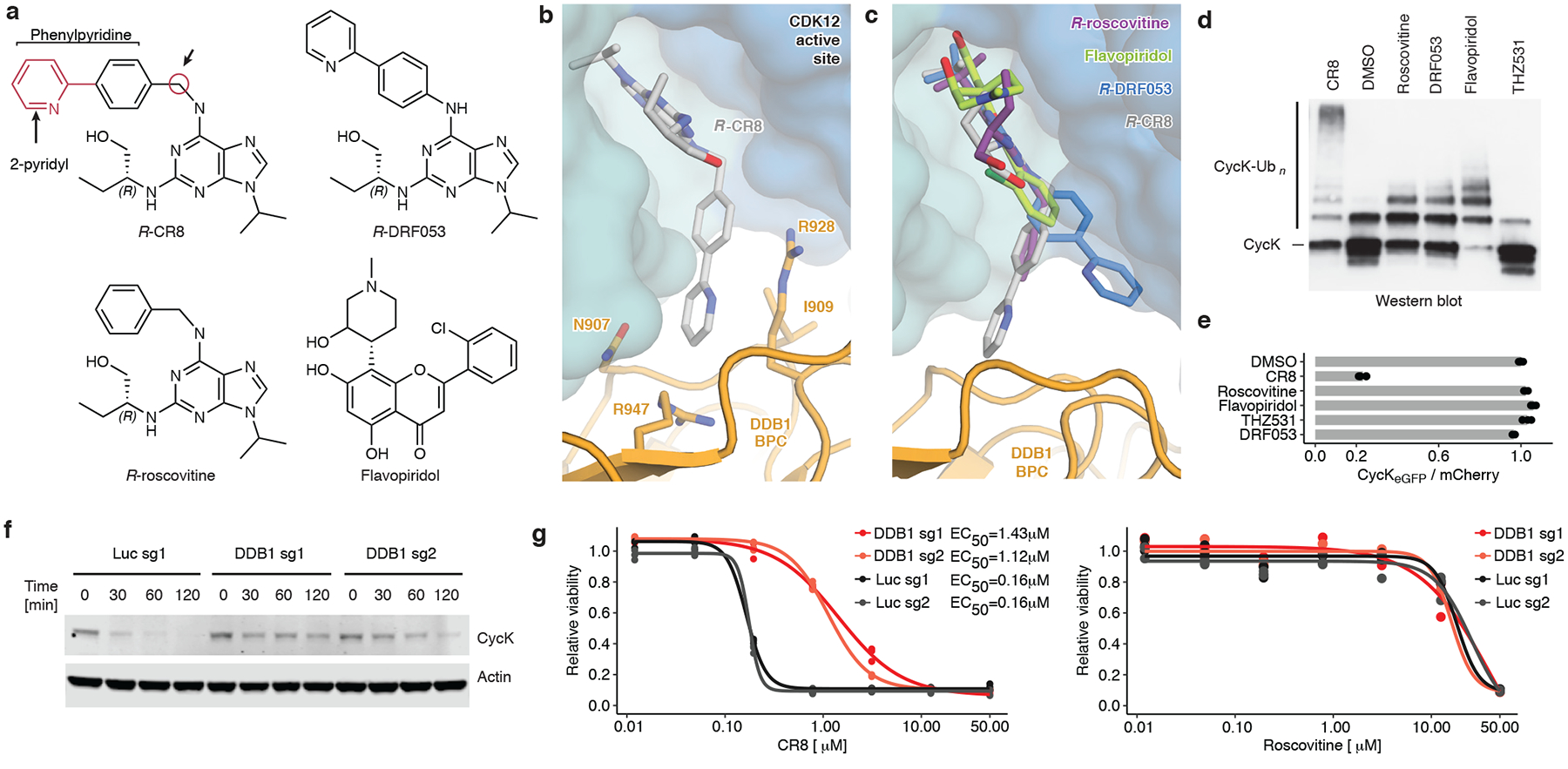
Molecular glue compounds induce protein-protein interactions that, in the context of a ubiquitin ligase, lead to protein degradation1. Unlike traditional enzyme inhibitors, these molecular glue degraders act substoichiometrically to catalyse the rapid depletion of previously inaccessible targets2. They are clinically effective and highly sought-after, but have thus far only been discovered serendipitously. Here, through systematically mining databases for correlations between the cytotoxicity of 4,518 clinical and preclinical small molecules and the expression levels of E3 ligase components across hundreds of human cancer cell lines3-5, we identify CR8-a cyclin-dependent kinase (CDK) inhibitor6-as a compound that acts as a molecular glue degrader. The CDK-bound form of CR8 has a solvent-exposed pyridyl moiety that induces the formation of a complex between CDK12-cyclin K and the CUL4 adaptor protein DDB1, bypassing the requirement for a substrate receptor and presenting cyclin K for ubiquitination and degradation. Our studies demonstrate that chemical alteration of surface-exposed moieties can confer gain-of-function glue properties to an inhibitor, and we propose this as a broader strategy through which target-binding molecules could be converted into molecular glues.
Conflict of interest statement
Competing interests
B.L.E. has received research funding from Celgene and Deerfield. He has received consulting fees from GRAIL, and he serves on the scientific advisory boards for and holds equity in Skyhawk Therapeutics and Exo Therapeutics. E.S.F. is a founder and/or member of the scientific advisory board (SAB), and equity holder of C4 Therapeutics and Civetta Therapeutics and a consultant to Novartis, AbbVie and Pfizer. N.H.T receives funding from the Novartis Research Foundation and is an SAB member of Monte Rosa Therapeutics. The Fischer lab receives or has received research funding from Novartis, Deerfield and Astellas. S.F. has had a consulting or advisory role, received honoraria, research funding, and/or travel/accommodation expenses funding from the following for-profit companies: Bayer, Roche, Amgen, Eli Lilly, PharmaMar, AstraZeneca, and Pfizer. R.B. is now an employee of Monte Rosa Therapeutics. S.M.C and T.R.G receive research funding from Bayer HealthCare. T.R.G. was formerly a consultant and equity holder in Foundation Medicine (acquired by Roche). T.R.G. also is a consultant to GlaxoSmithKline and is a founder of Sherlock Biosciences.
Figures
Comment in
-
Prospecting for molecular glues.Nat Chem Biol. 2020 Nov;16(11):1157-1158. doi: 10.1038/s41589-020-0620-z. Nat Chem Biol. 2020. PMID: 32747810 No abstract available.
References
-
- Corsello SM et al. Non-oncology drugs are a source of previously unappreciated anti-cancer activity. Biorxiv (2019). doi: 10.1101/730119 - DOI
Publication types
MeSH terms
Substances
Grants and funding
- R35 CA253125/CA/NCI NIH HHS/United States
- KL2 TR002542/TR/NCATS NIH HHS/United States
- P50 CA206963/CA/NCI NIH HHS/United States
- R01 HL082945/HL/NHLBI NIH HHS/United States
- T32 GM007753/GM/NIGMS NIH HHS/United States
- P01 CA108631/CA/NCI NIH HHS/United States
- HHMI/Howard Hughes Medical Institute/United States
- R01 CA214608/CA/NCI NIH HHS/United States
- R01 CA218278/CA/NCI NIH HHS/United States
- 29036/CRUK_/Cancer Research UK/United Kingdom
- P01 CA066996/CA/NCI NIH HHS/United States
- K08 CA230220/CA/NCI NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
