

Mouse models of neutropenia reveal progenitor-stage-specific defects
- PMID: 32494068
- PMCID: PMC8041154
- DOI: 10.1038/s41586-020-2227-7
Mouse models of neutropenia reveal progenitor-stage-specific defects
Abstract
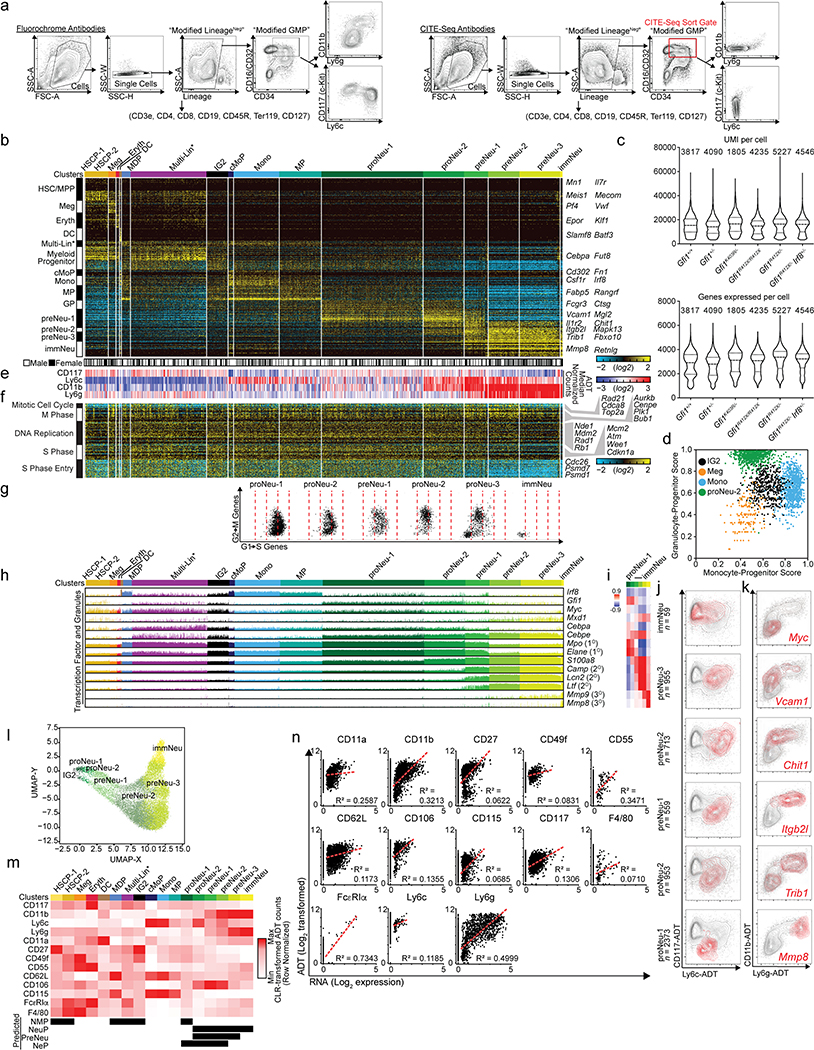
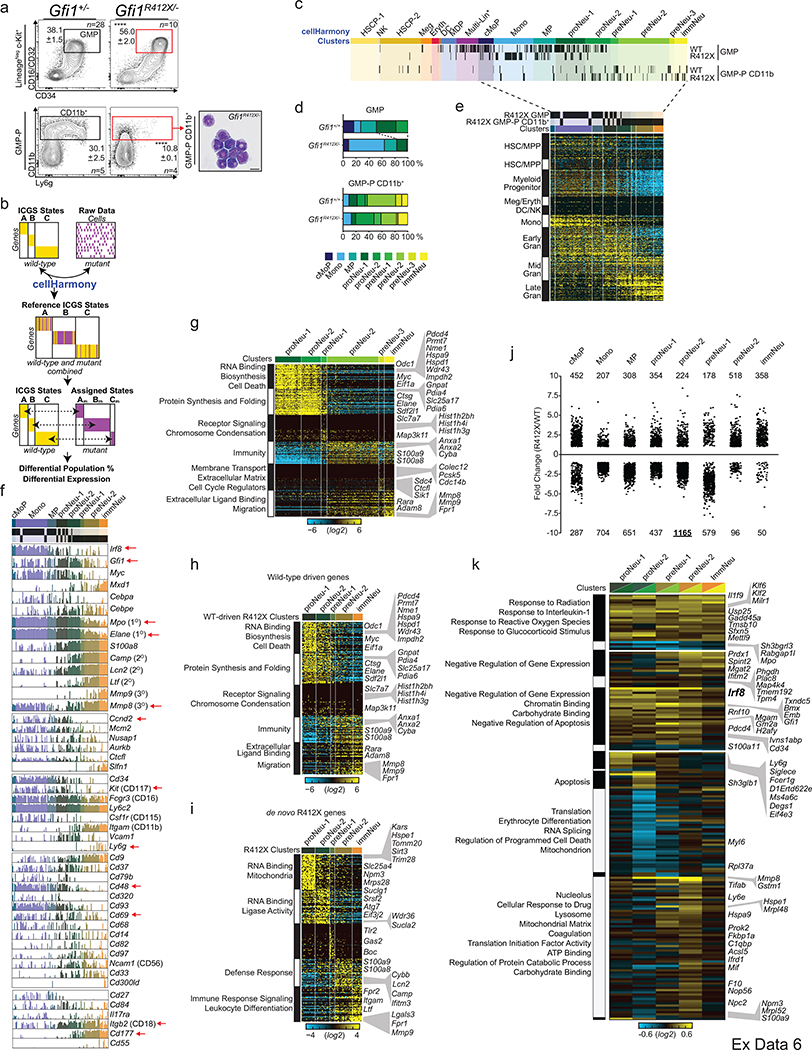
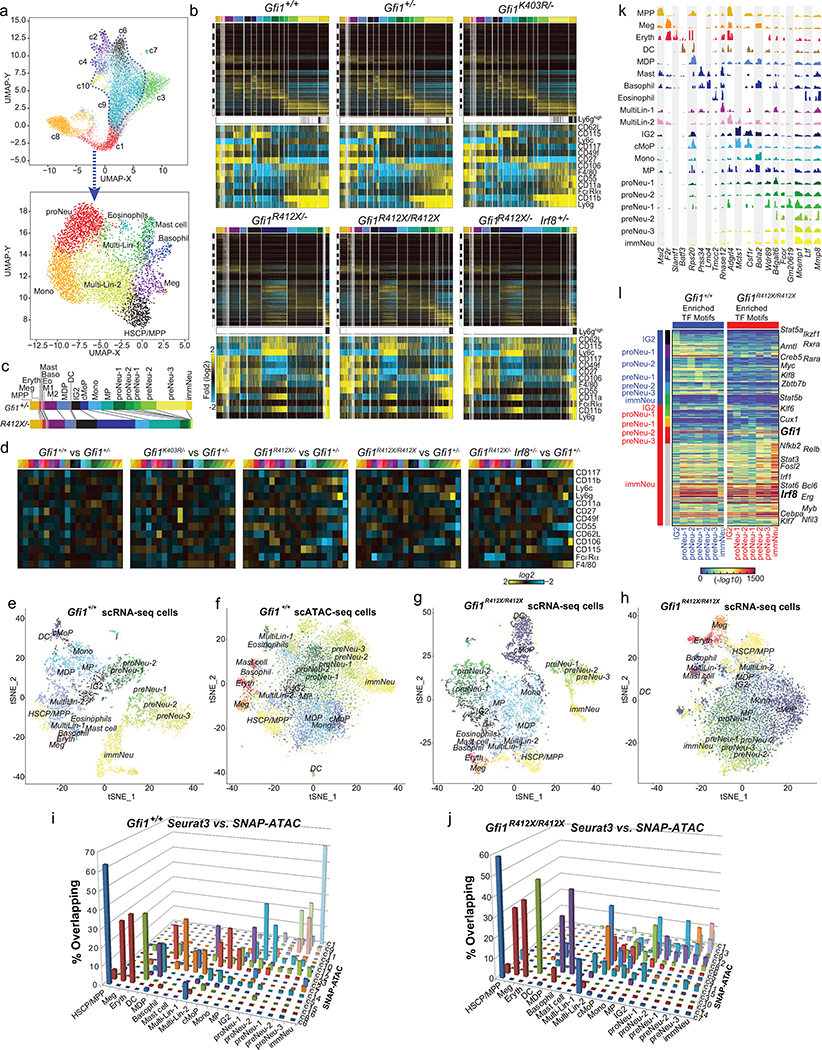
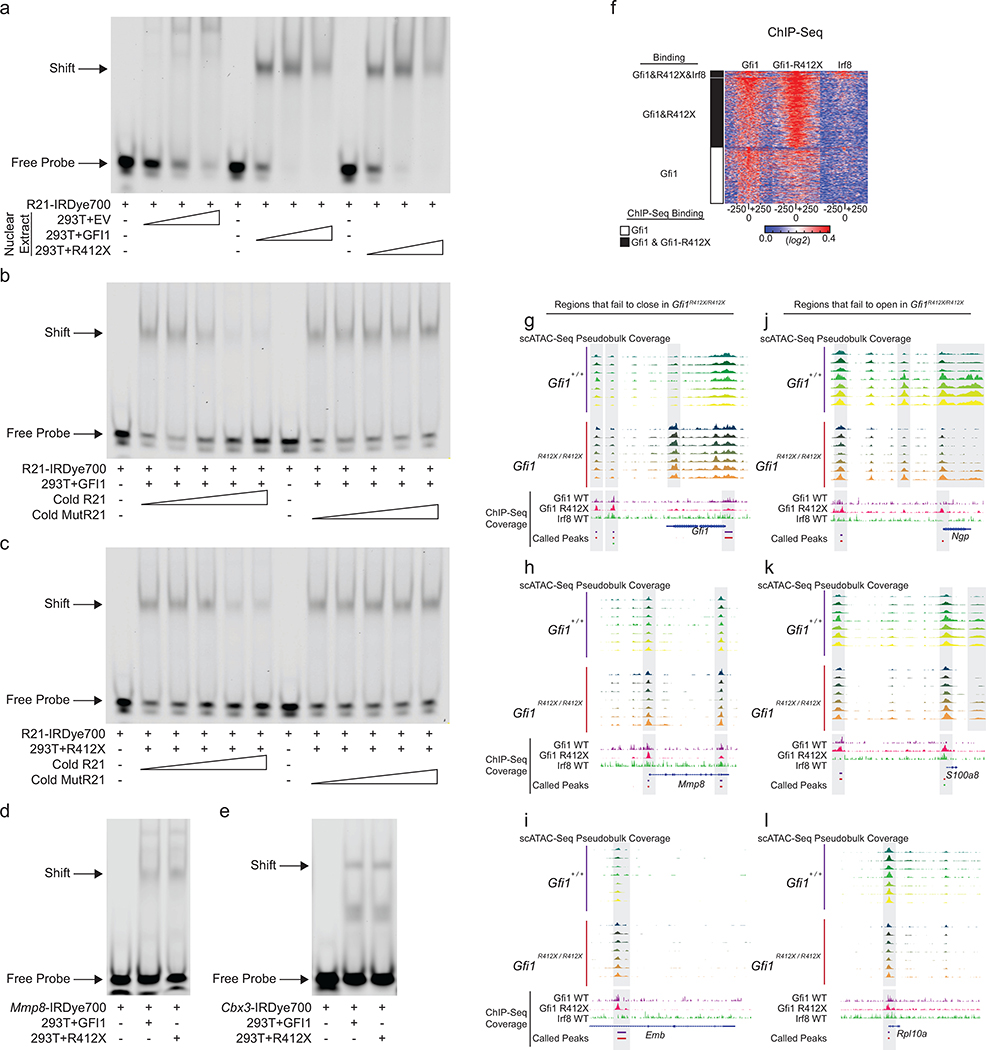
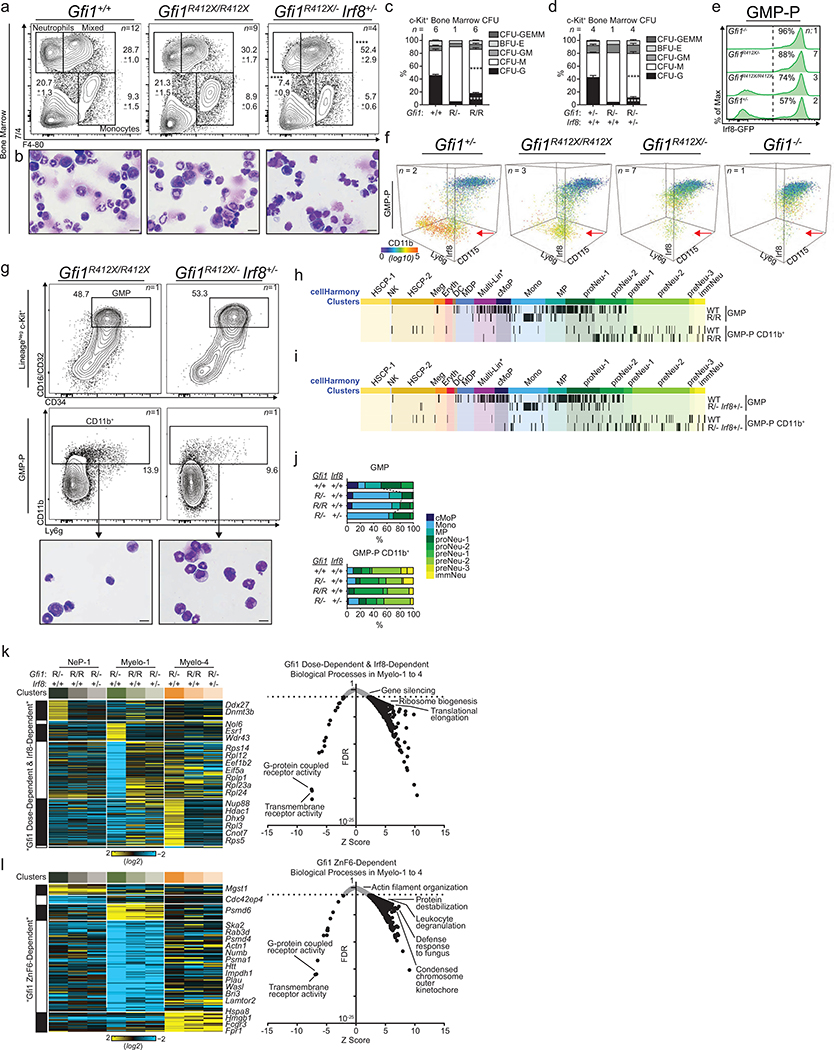
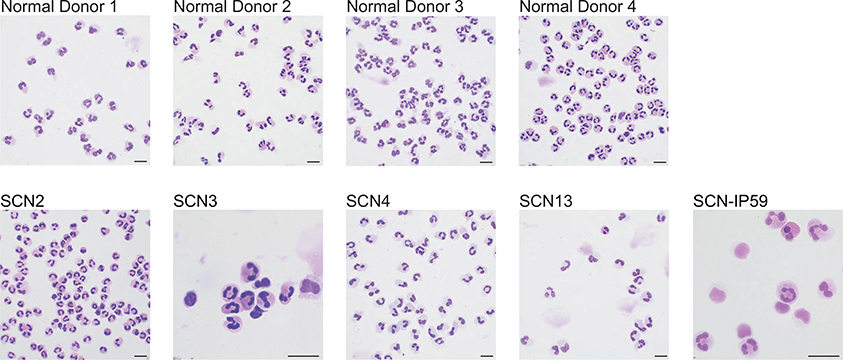
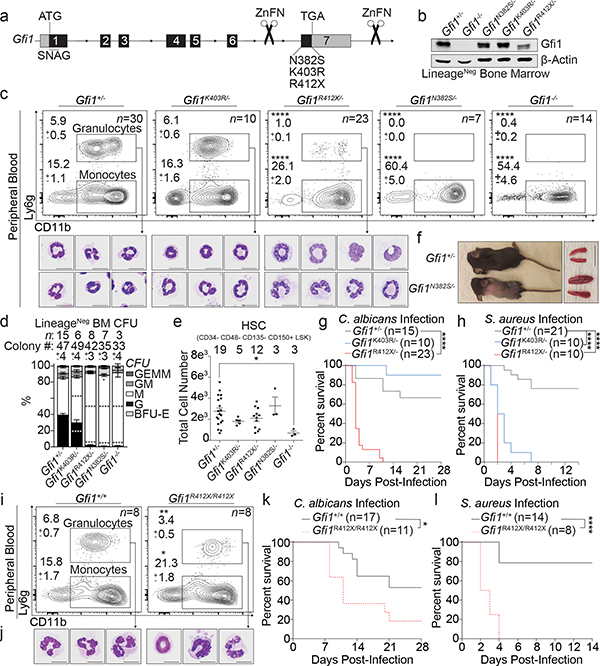
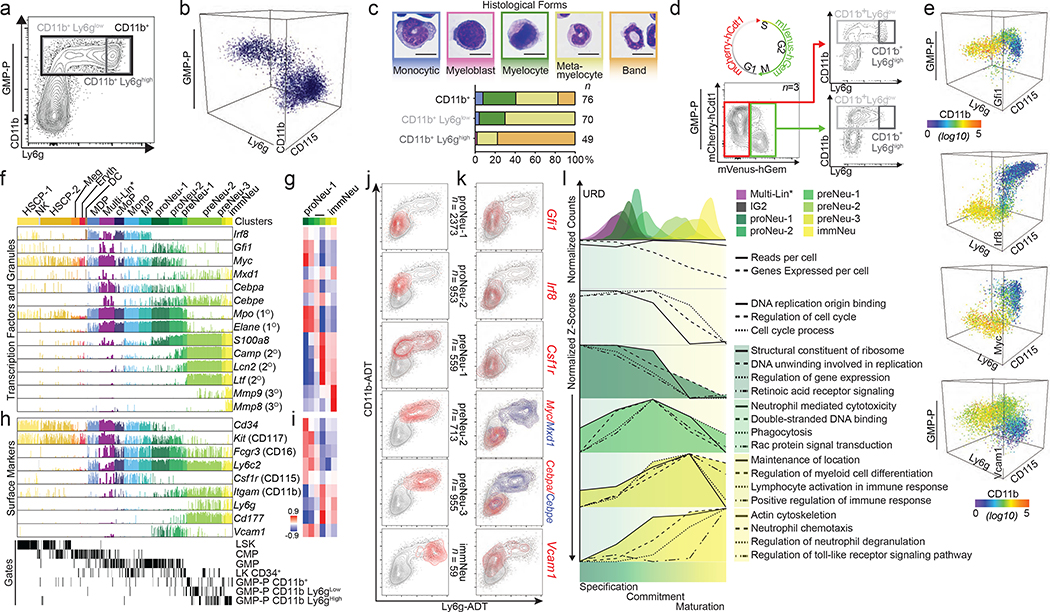
Advances in genetics and sequencing have identified a plethora of disease-associated and disease-causing genetic alterations. To determine causality between genetics and disease, accurate models for molecular dissection are required; however, the rapid expansion of transcriptional populations identified through single-cell analyses presents a major challenge for accurate comparisons between mutant and wild-type cells. Here we generate mouse models of human severe congenital neutropenia (SCN) using patient-derived mutations in the GFI1 transcription factor. To determine the effects of SCN mutations, we generated single-cell references for granulopoietic genomic states with linked epitopes1, aligned mutant cells to their wild-type equivalents and identified differentially expressed genes and epigenetic loci. We find that GFI1-target genes are altered sequentially, as cells go through successive states of differentiation. These insights facilitated the genetic rescue of granulocytic specification but not post-commitment defects in innate immune effector function, and underscore the importance of evaluating the effects of mutations and therapy within each relevant cell state.
Conflict of interest statement
The authors declare the following competing interests: DEM is currently employed by Eli Lilly and Company and KLN is employed by BioLegend Inc. The remaining authors declare no competing interests.
Figures
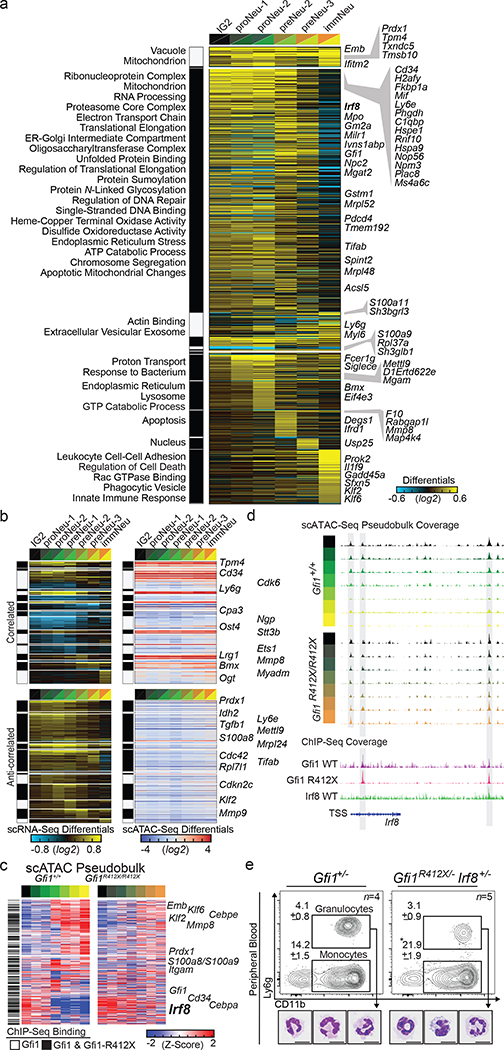
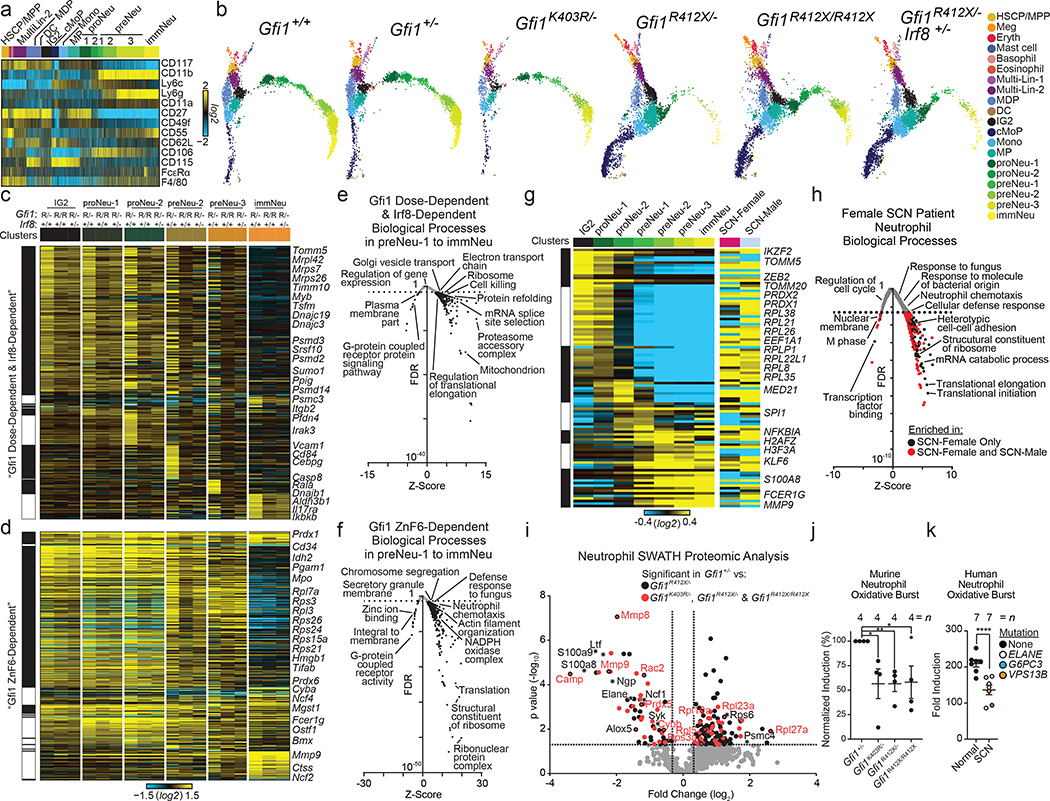
References
-
- Gilbert SF Developmental Biology, 6th edition. (Sinauer Associates, 2000).
-
- An interactive online viewer of murine hematopoietic gene expression, <http://www.altanalyze.org/ICGS/Neutrophil/Viewer.php> (
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
