

Vascular Calcification: An Important Understanding in Nephrology
- PMID: 32494148
- PMCID: PMC7229867
- DOI: 10.2147/VHRM.S242685
Vascular Calcification: An Important Understanding in Nephrology
Abstract
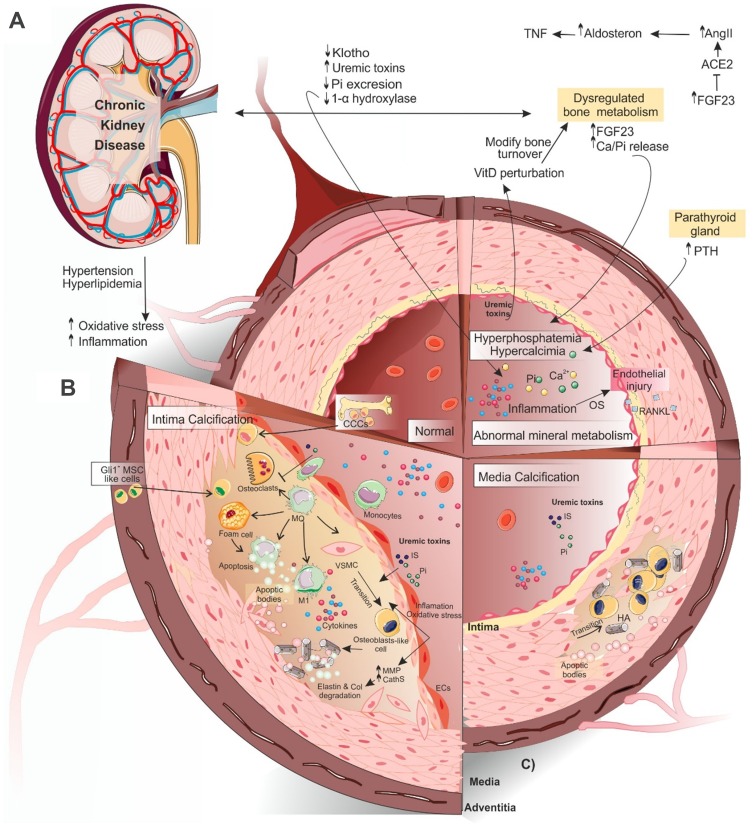
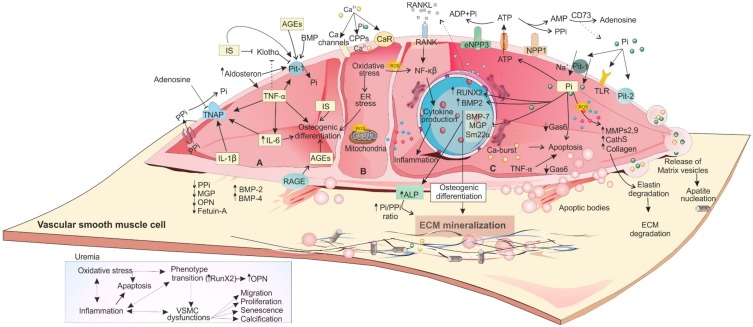
Vascular calcification (VC) is a life-threatening state in chronic kidney disease (CKD). High cardiovascular mortality and morbidity of CKD cases may root from medial VC promoted by hyperphosphatemia. Vascular calcification is an active, highly regulated, and complex biological process that is mediated by genetics, epigenetics, dysregulated form of matrix mineral metabolism, hormones, and the activation of cellular signaling pathways. Moreover, gut microbiome as a source of uremic toxins (eg, phosphate, advanced glycation end products and indoxyl-sulfate) can be regarded as a potential contributor to VC in CKD. Here, an update on different cellular and molecular processes involved in VC in CKD is discussed to elucidate the probable therapeutic pathways in the future.
Keywords: CKD; calcification; chronic kidney disease; hyperphosphatemia; uremia; uremic toxins.
© 2020 Zununi Vahed et al.
Conflict of interest statement
The authors declare that they have no conflict of interest regarding this work.
Figures
Similar articles
-
Contribution of Gut Microbiota-Derived Uremic Toxins to the Cardiovascular System Mineralization.Toxins (Basel). 2021 Apr 10;13(4):274. doi: 10.3390/toxins13040274. Toxins (Basel). 2021. PMID: 33920096 Free PMC article. Review.
-
Uremic Vascular Calcification: The Pathogenic Roles and Gastrointestinal Decontamination of Uremic Toxins.Toxins (Basel). 2020 Dec 21;12(12):812. doi: 10.3390/toxins12120812. Toxins (Basel). 2020. PMID: 33371477 Free PMC article. Review.
-
Effects of Chronic Kidney Disease and Uremic Toxins on Extracellular Vesicle Biology.Toxins (Basel). 2020 Dec 21;12(12):811. doi: 10.3390/toxins12120811. Toxins (Basel). 2020. PMID: 33371311 Free PMC article. Review.
-
The activin receptor is stimulated in the skeleton, vasculature, heart, and kidney during chronic kidney disease.Kidney Int. 2018 Jan;93(1):147-158. doi: 10.1016/j.kint.2017.06.016. Epub 2017 Aug 23. Kidney Int. 2018. PMID: 28843411 Free PMC article.
-
Phosphate is a vascular toxin.Pediatr Nephrol. 2013 Apr;28(4):583-93. doi: 10.1007/s00467-012-2347-x. Epub 2012 Nov 17. Pediatr Nephrol. 2013. PMID: 23161206 Review.
Cited by
-
Hypoxia-inducible factor signaling in vascular calcification in chronic kidney disease patients.J Nephrol. 2022 Dec;35(9):2205-2213. doi: 10.1007/s40620-022-01432-8. Epub 2022 Oct 8. J Nephrol. 2022. PMID: 36208406 Review.
-
Higher serum trimethylamine-N-oxide levels are associated with increased abdominal aortic calcification in hemodialysis patients.Ren Fail. 2022 Dec;44(1):2019-2027. doi: 10.1080/0886022X.2022.2145971. Ren Fail. 2022. PMID: 36384389 Free PMC article.
-
Contribution of Gut Microbiota-Derived Uremic Toxins to the Cardiovascular System Mineralization.Toxins (Basel). 2021 Apr 10;13(4):274. doi: 10.3390/toxins13040274. Toxins (Basel). 2021. PMID: 33920096 Free PMC article. Review.
-
Accumulation of Advanced Glycation End-Products in the Body and Dietary Habits.Nutrients. 2022 Sep 25;14(19):3982. doi: 10.3390/nu14193982. Nutrients. 2022. PMID: 36235635 Free PMC article. Review.
-
Effects of sevelamer carbonate versus calcium acetate on vascular calcification, inflammation, and endothelial dysfunction in chronic kidney disease.Clin Transl Sci. 2022 Feb;15(2):353-360. doi: 10.1111/cts.13151. Epub 2021 Oct 2. Clin Transl Sci. 2022. PMID: 34599865 Free PMC article.
References
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Medical
