

Integrated analysis of the molecular pathogenesis of FDXR-associated disease
- PMID: 32499495
- PMCID: PMC7272433
- DOI: 10.1038/s41419-020-2637-3
Integrated analysis of the molecular pathogenesis of FDXR-associated disease
Abstract
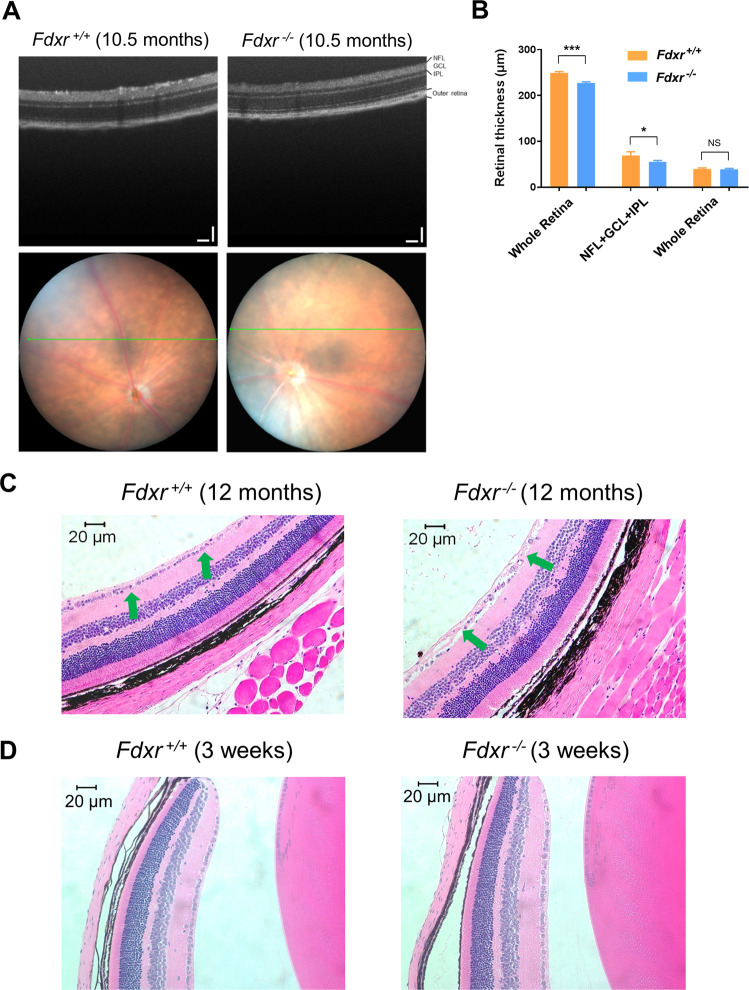
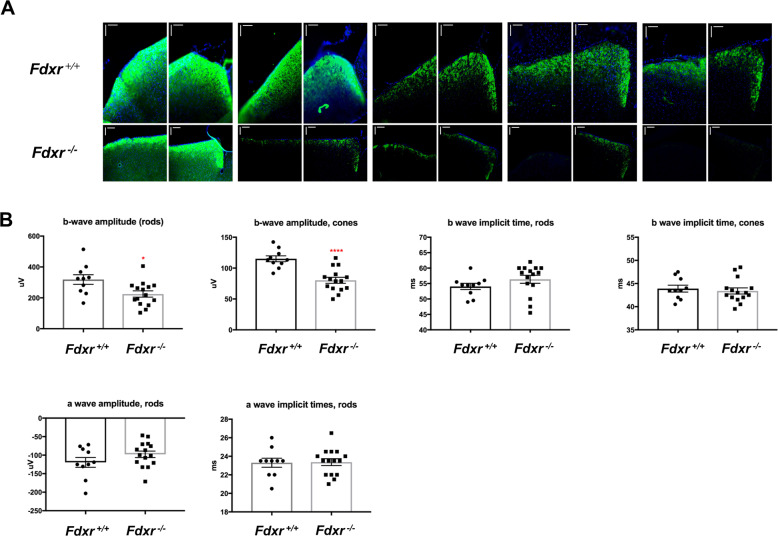
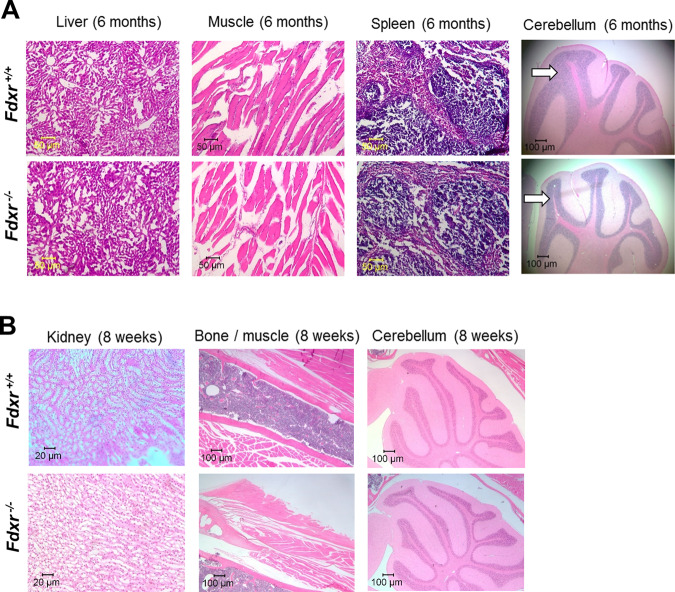
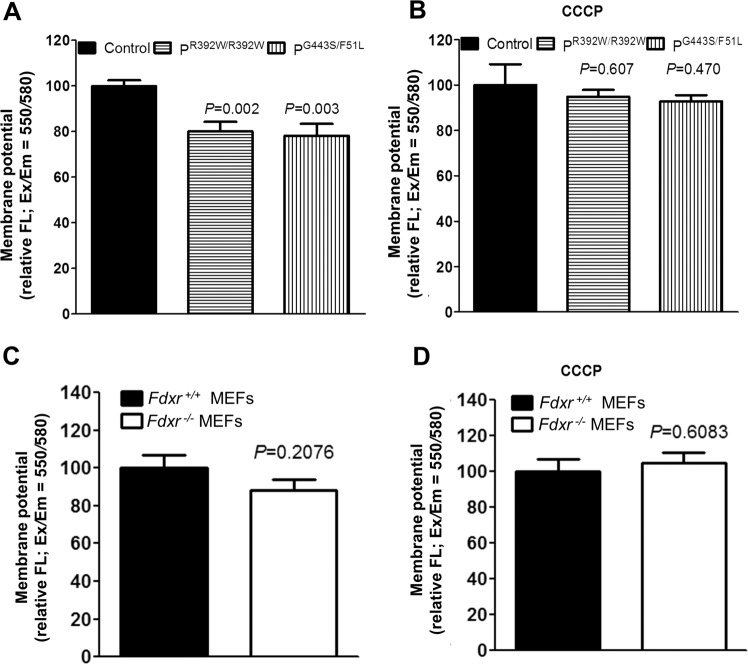
The mitochondrial flavoprotein ferredoxin reductase (FDXR) is required for biogenesis of iron-sulfur clusters and for steroidogenesis. Iron-sulfur (Fe-S) clusters are ubiquitous cofactors essential to various cellular processes, and an increasing number of disorders are associated with disruptions in the synthesis of Fe-S clusters. Our previous studies have demonstrated that hypomorphic mutations in FDXR cause a novel mitochondriopathy and optic atrophy in humans and mice, attributed in part to reduced function of the electron transport chain (ETC) as well as elevated production of reactive oxygen species (ROS). Inflammation and peripheral neuropathy are also hallmarks of this disease. In this paper, we demonstrate that FDXR mutation leads to significant optic transport defects that are likely to underlie optic atrophy, a major clinical presentation in FDXR patients, as well as a neurodegenerative loss of cells in the central nervous system (CNS). Molecular analysis indicates that FDXR mutation also leads to mitochondrial iron overload and an associated depolarization of the mitochondrial membrane, further supporting the hypothesis that FDXR mutations cause neurodegeneration by affecting FDXR's critical role in iron homeostasis.
Conflict of interest statement
The authors declare that they have no conflict of interest.
Figures


References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Miscellaneous
