

Whole-genome sequencing of a sporadic primary immunodeficiency cohort
- PMID: 32499645
- PMCID: PMC7334047
- DOI: 10.1038/s41586-020-2265-1
Whole-genome sequencing of a sporadic primary immunodeficiency cohort
Erratum in
-
Publisher Correction: Whole-genome sequencing of a sporadic primary immunodeficiency cohort.Nature. 2020 Aug;584(7819):E2. doi: 10.1038/s41586-020-2556-6. Nature. 2020. PMID: 32678341
Abstract
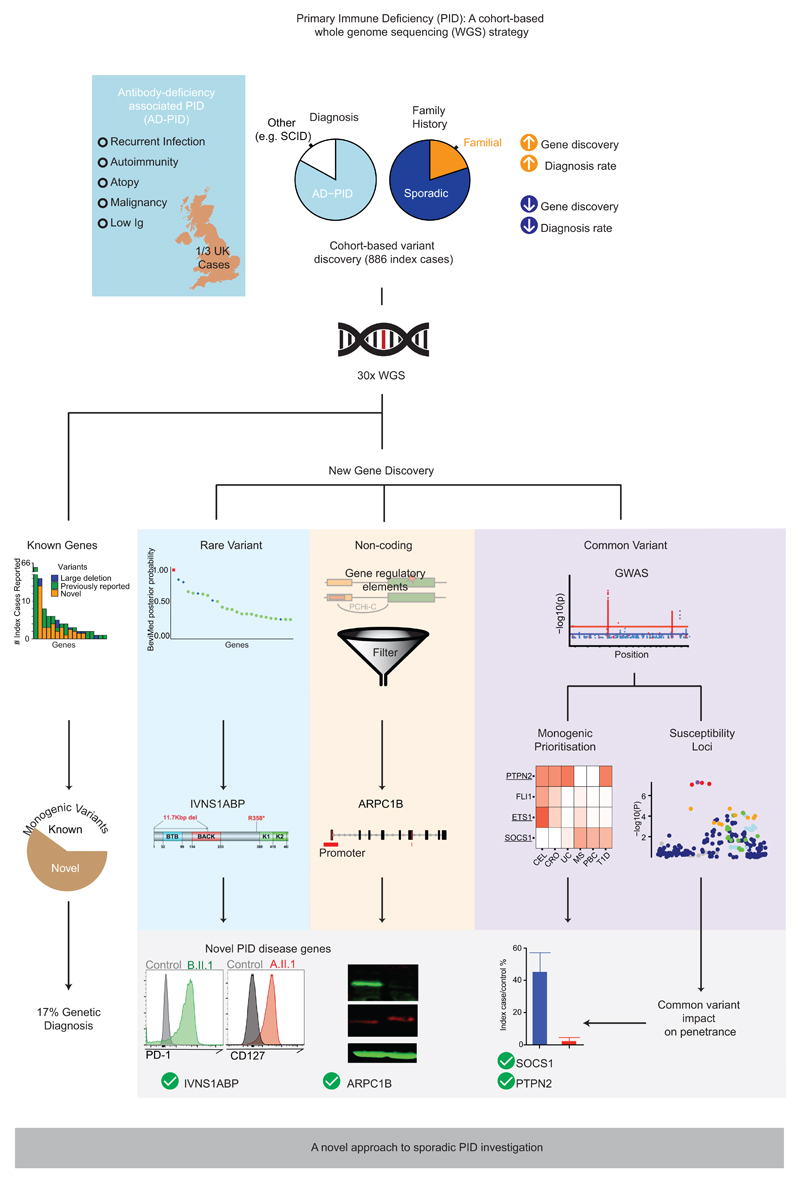
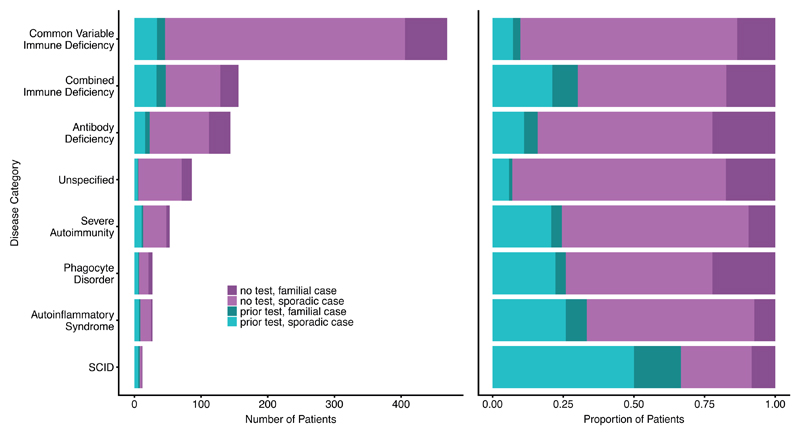
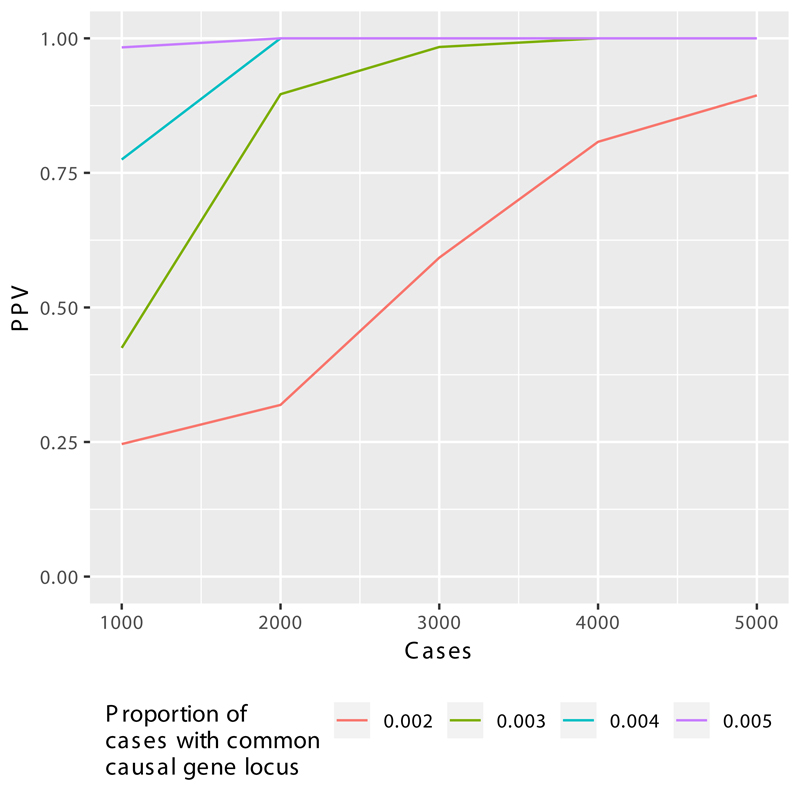
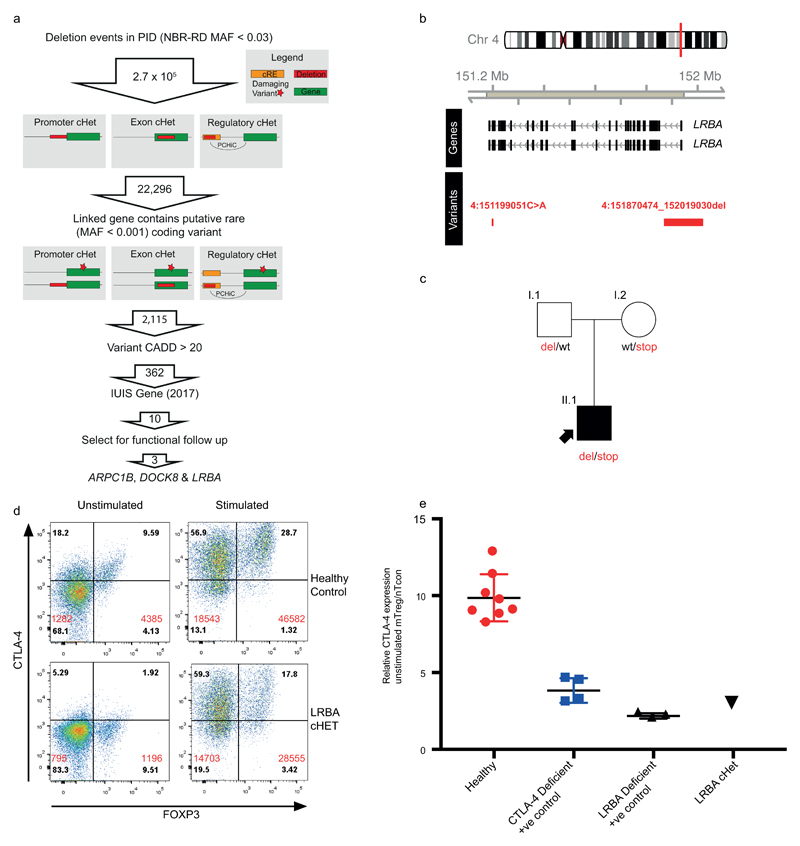
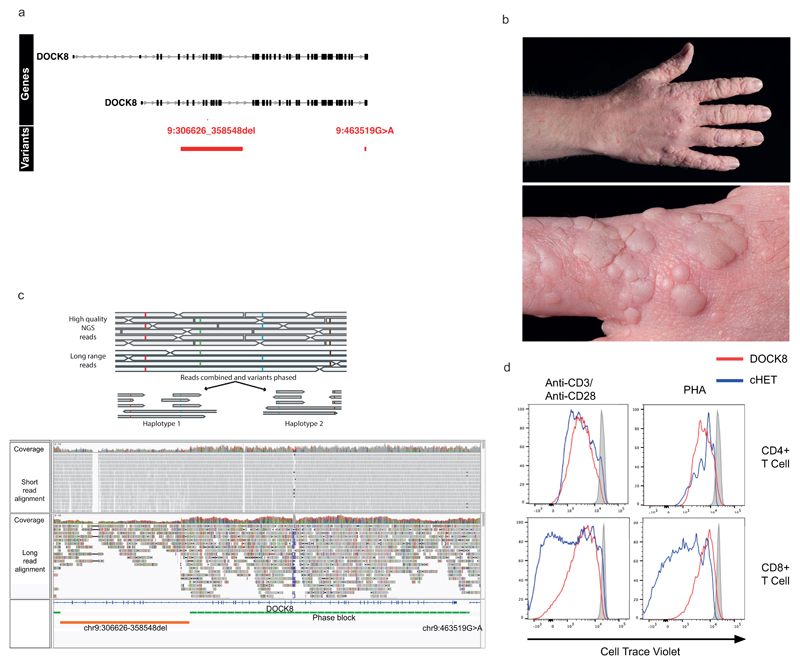
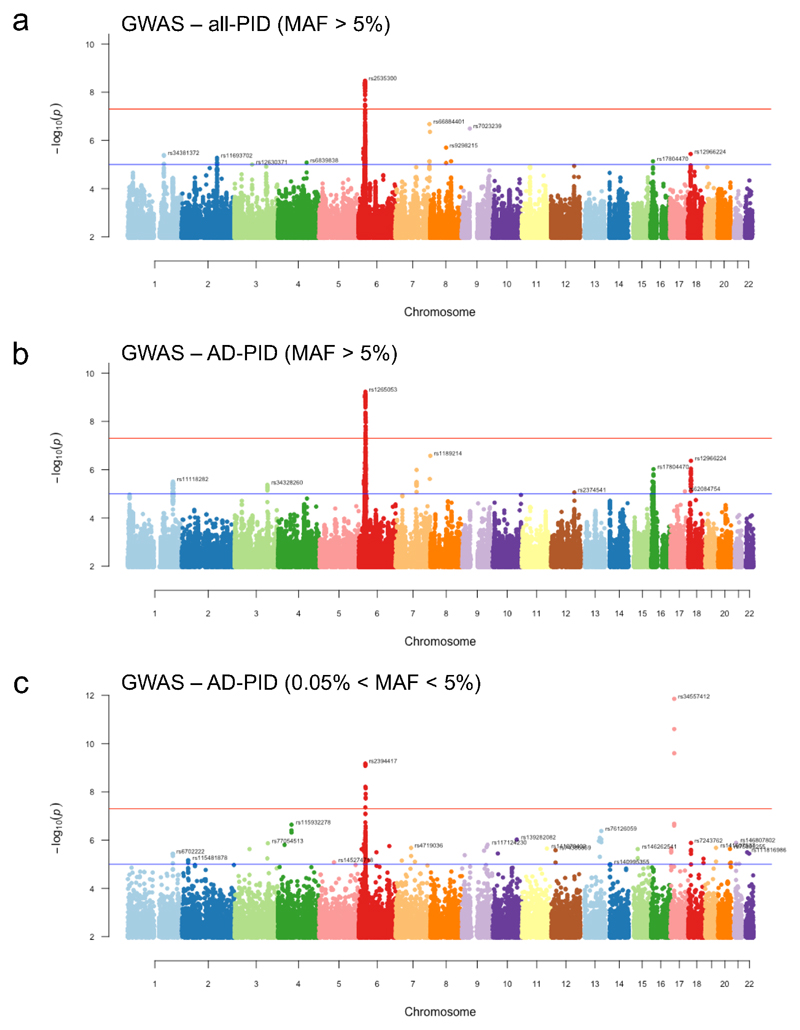
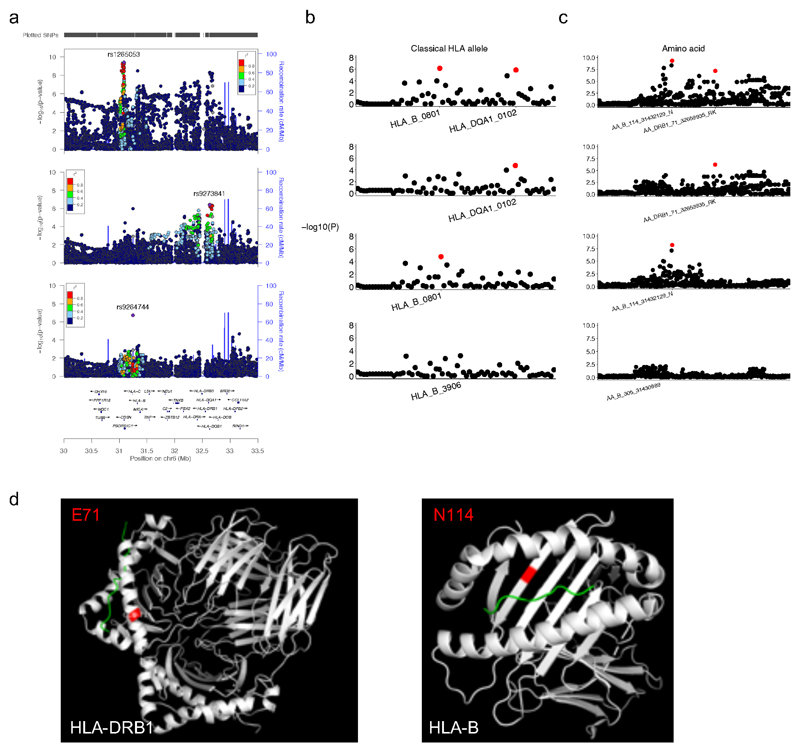
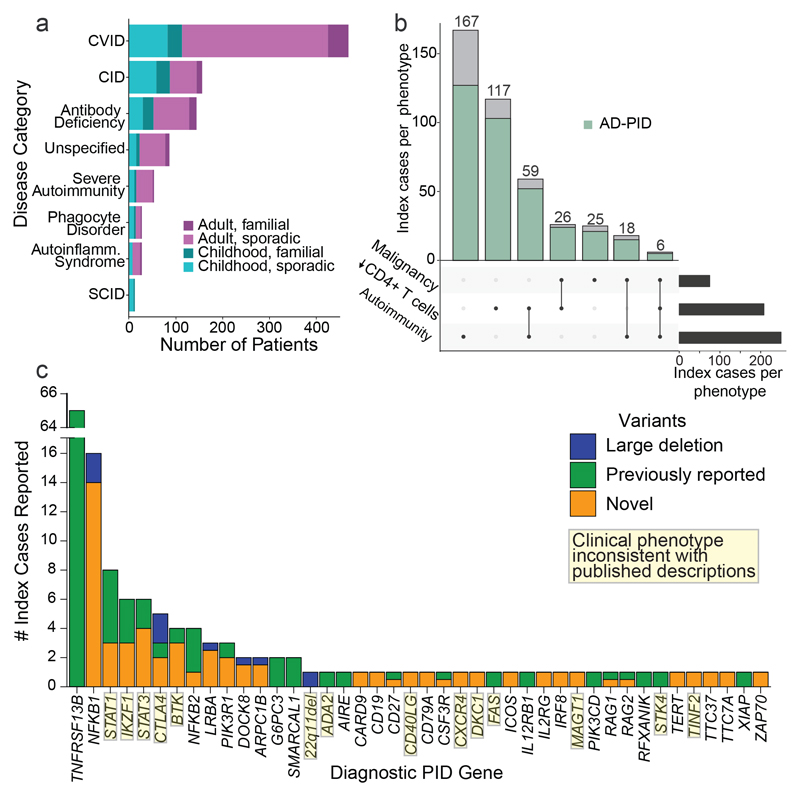
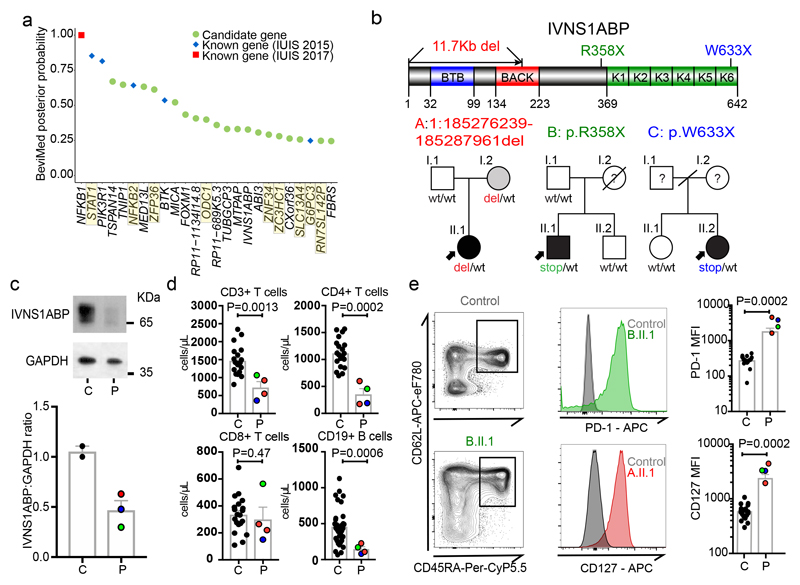
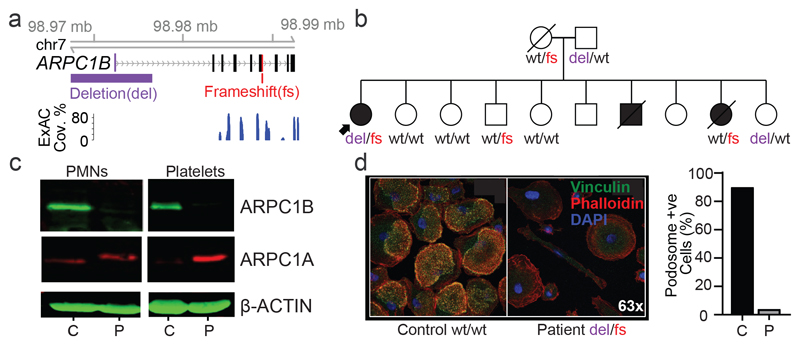
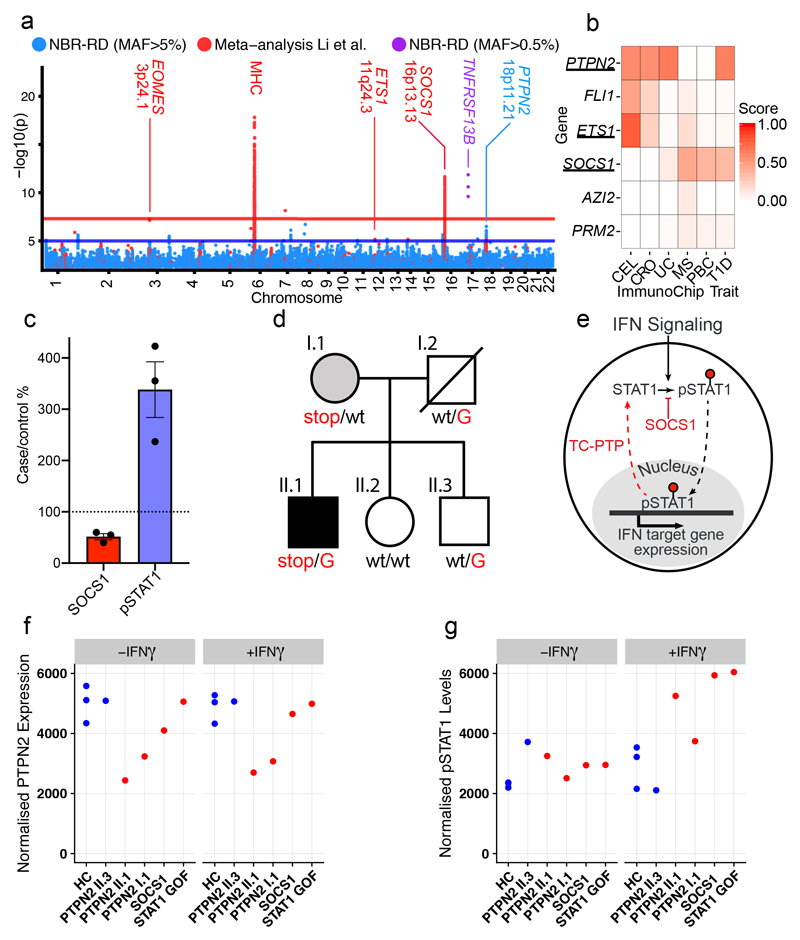
Primary immunodeficiency (PID) is characterized by recurrent and often life-threatening infections, autoimmunity and cancer, and it poses major diagnostic and therapeutic challenges. Although the most severe forms of PID are identified in early childhood, most patients present in adulthood, typically with no apparent family history and a variable clinical phenotype of widespread immune dysregulation: about 25% of patients have autoimmune disease, allergy is prevalent and up to 10% develop lymphoid malignancies1-3. Consequently, in sporadic (or non-familial) PID genetic diagnosis is difficult and the role of genetics is not well defined. Here we address these challenges by performing whole-genome sequencing in a large PID cohort of 1,318 participants. An analysis of the coding regions of the genome in 886 index cases of PID found that disease-causing mutations in known genes that are implicated in monogenic PID occurred in 10.3% of these patients, and a Bayesian approach (BeviMed4) identified multiple new candidate PID-associated genes, including IVNS1ABP. We also examined the noncoding genome, and found deletions in regulatory regions that contribute to disease causation. In addition, we used a genome-wide association study to identify loci that are associated with PID, and found evidence for the colocalization of-and interplay between-novel high-penetrance monogenic variants and common variants (at the PTPN2 and SOCS1 loci). This begins to explain the contribution of common variants to the variable penetrance and phenotypic complexity that are observed in PID. Thus, using a cohort-based whole-genome-sequencing approach in the diagnosis of PID can increase diagnostic yield and further our understanding of the key pathways that influence immune responsiveness in humans.
Conflict of interest statement
The authors declare no competing financial interests.
Figures
References
-
- Gathmann B, et al. Clinical picture and treatment of 2212 patients with common variable immunodeficiency. J Allergy Clin Immunol. 2014;134:116–126.e11. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- SP/12/12/29836/BHF_/British Heart Foundation/United Kingdom
- G84/6443/MRC_/Medical Research Council/United Kingdom
- 28051/CRUK_/Cancer Research UK/United Kingdom
- 26988/CRUK_/Cancer Research UK/United Kingdom
- MR/S001190/1/MRC_/Medical Research Council/United Kingdom
- MC_UU_00016/15/MRC_/Medical Research Council/United Kingdom
- MR/K020919/1/MRC_/Medical Research Council/United Kingdom
- MC_UU_12009/16/MRC_/Medical Research Council/United Kingdom
- 104807/WT_/Wellcome Trust/United Kingdom
- MC_UU_12015/2/MRC_/Medical Research Council/United Kingdom
- MC_UP_1102/20/MRC_/Medical Research Council/United Kingdom
- 201250/WT_/Wellcome Trust/United Kingdom
- 091157/WT_/Wellcome Trust/United Kingdom
- MR/S021329/1/MRC_/Medical Research Council/United Kingdom
- FS/18/53/33863/BHF_/British Heart Foundation/United Kingdom
- MR/P02002X/1/MRC_/Medical Research Council/United Kingdom
- CH/1992001/6764/BHF_/British Heart Foundation/United Kingdom
- RP-2016-07-019/DH_/Department of Health/United Kingdom
- 203141/WT_/Wellcome Trust/United Kingdom
- MR/L006197/1/MRC_/Medical Research Council/United Kingdom
- 27723/CRUK_/Cancer Research UK/United Kingdom
- MR/L006340/1/MRC_/Medical Research Council/United Kingdom
- 202747/Z/16/Z/WT_/Wellcome Trust/United Kingdom
- 29034/CRUK_/Cancer Research UK/United Kingdom
- 204798/Z/16/Z/WT_/Wellcome Trust/United Kingdom
- MR/L019027/1/MRC_/Medical Research Council/United Kingdom
- 107212/WT_/Wellcome Trust/United Kingdom
- 100140/WT_/Wellcome Trust/United Kingdom
- MC_UU_00006/2/MRC_/Medical Research Council/United Kingdom
- 27125/CRUK_/Cancer Research UK/United Kingdom
- MR/K02342X/1/MRC_/Medical Research Council/United Kingdom
- MC_PC_18030/MRC_/Medical Research Council/United Kingdom
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
