

Branched evolution and genomic intratumor heterogeneity in the pathogenesis of cutaneous T-cell lymphoma
- PMID: 32502269
- PMCID: PMC7284100
- DOI: 10.1182/bloodadvances.2020001441
Branched evolution and genomic intratumor heterogeneity in the pathogenesis of cutaneous T-cell lymphoma
Abstract
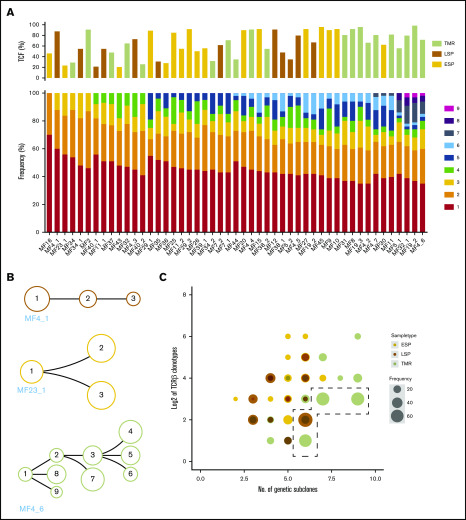
Mycosis fungoides (MF) is a slowly progressive cutaneous T-cell lymphoma (CTCL) for which there is no cure. In the early plaque stage, the disease is indolent, but development of tumors heralds an increased risk of metastasis and death. Previous research into the genomic landscape of CTCL revealed a complex pattern of >50 driver mutations implicated in more than a dozen signaling pathways. However, the genomic mechanisms governing disease progression and treatment resistance remain unknown. Building on our previous discovery of the clonotypic heterogeneity of MF, we hypothesized that this lymphoma does not progress in a linear fashion as currently thought but comprises heterogeneous mutational subclones. We sequenced exomes of 49 cases of MF and identified 28 previously unreported putative driver genes. MF exhibited extensive intratumoral heterogeneity (ITH) of a median of 6 subclones showing a branched phylogenetic relationship pattern. Stage progression was correlated with an increase in ITH and redistribution of mutations from stem to clades. The pattern of clonal driver mutations was highly variable, with no consistent mutations among patients. Similar intratumoral heterogeneity was detected in leukemic CTCL (Sézary syndrome). Based on these findings, we propose a model of MF pathogenesis comprising divergent evolution of cancer subclones and discuss how ITH affects the efficacy of targeted drug therapies and immunotherapies for CTCL.
© 2020 by The American Society of Hematology.
Conflict of interest statement
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Figures







References
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Medical
