

Exploring the genomic and proteomic variations of SARS-CoV-2 spike glycoprotein: A computational biology approach
- PMID: 32502733
- PMCID: PMC7266584
- DOI: 10.1016/j.meegid.2020.104389
Exploring the genomic and proteomic variations of SARS-CoV-2 spike glycoprotein: A computational biology approach
Abstract
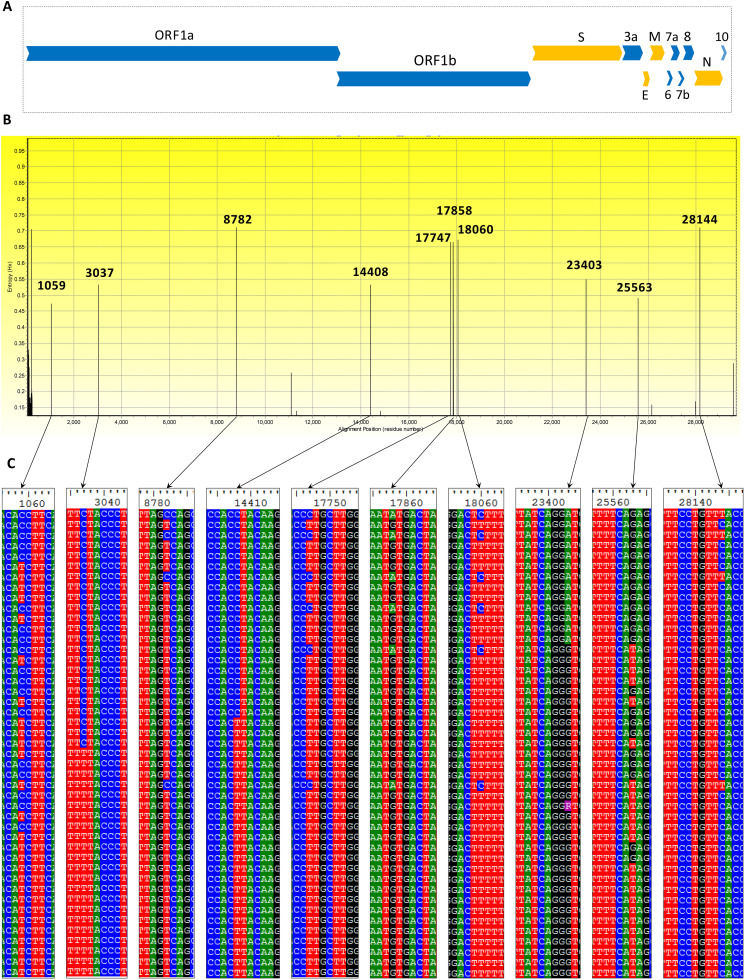
The newly identified SARS-CoV-2 has now been reported from around 185 countries with more than a million confirmed human cases including more than 120,000 deaths. The genomes of SARS-COV-2 strains isolated from different parts of the world are now available and the unique features of constituent genes and proteins need to be explored to understand the biology of the virus. Spike glycoprotein is one of the major targets to be explored because of its role during the entry of coronaviruses into host cells. We analyzed 320 whole-genome sequences and 320 spike protein sequences of SARS-CoV-2 using multiple sequence alignment. In this study, 483 unique variations have been identified among the genomes of SARS-CoV-2 including 25 nonsynonymous mutations and one deletion in the spike (S) protein. Among the 26 variations detected in S, 12 variations were located at the N-terminal domain (NTD) and 6 variations at the receptor-binding domain (RBD) which might alter the interaction of S protein with the host receptor angiotensin-converting enzyme 2 (ACE2). Besides, 22 amino acid insertions were identified in the spike protein of SARS-CoV-2 in comparison with that of SARS-CoV. Phylogenetic analyses of spike protein revealed that Bat coronavirus have a close evolutionary relationship with circulating SARS-CoV-2. The genetic variation analysis data presented in this study can help a better understanding of SARS-CoV-2 pathogenesis. Based on results reported herein, potential inhibitors against S protein can be designed by considering these variations and their impact on protein structure.
Keywords: COVID-19; Genomic variants; SARS-CoV-2; Sequence analysis; Spike protein.
Copyright © 2020 Elsevier B.V. All rights reserved.
Conflict of interest statement
Declaration of Competing Interest The authors would like to declare that there is no known contending financial interests or personal relationships that could affect the work reported in this paper.
Figures





References
-
- Bosch B.J., Rossen J.W.A., Bartelink W., Zuurveen S.J., de Haan C.A.M., Duquerroy S., Boucher C.A.B., Rottier P.J.M. Coronavirus escape from heptad repeat 2 (HR2)-derived peptide entry inhibition as a result of mutations in the HR1 domain of the spike fusion protein. J. Virol. 2008;82:2580–2585. - PMC - PubMed
-
- Cárdenas-Conejo Y., Liñan-Rico A., Garcia-Rodriguez D.A., Centeno-Leija S., Serrano-Posada H. An exclusive 42 amino acid signature in pp1ab protein provides insights into the evolutive history of the 2019 novel human-pathogenic coronavirus (SARS-CoV2) J. Med. Virol. 2020;92:688–692. doi: 10.1002/jmv.25758. - DOI - PMC - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
