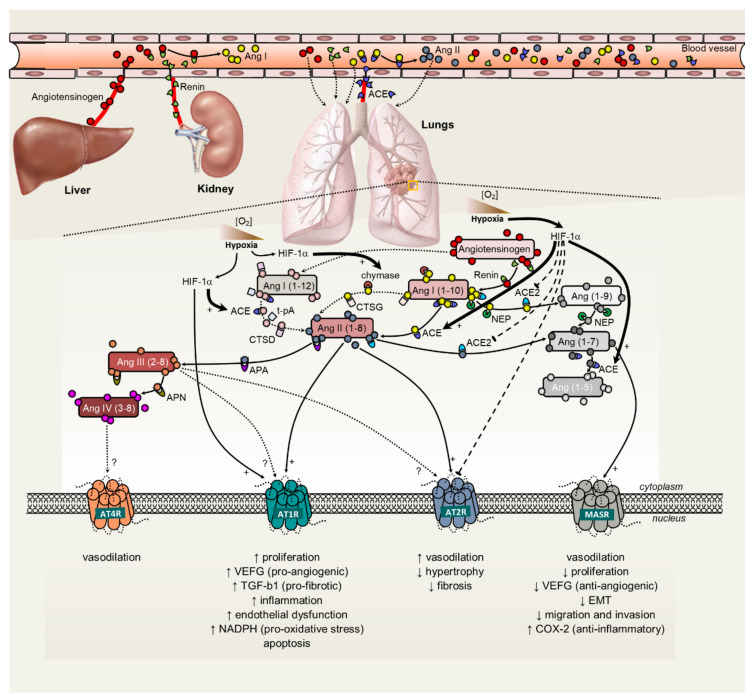
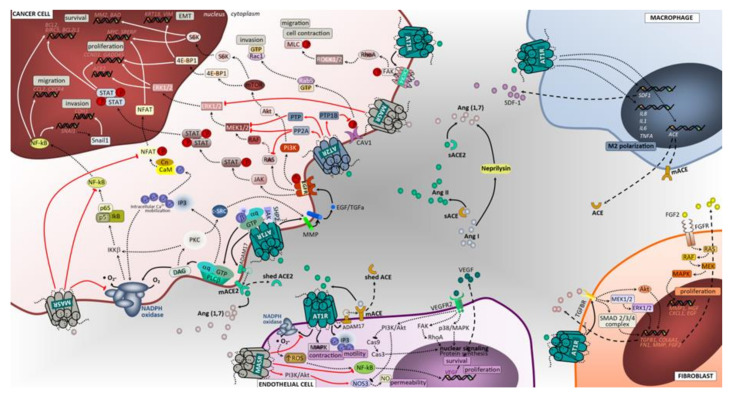
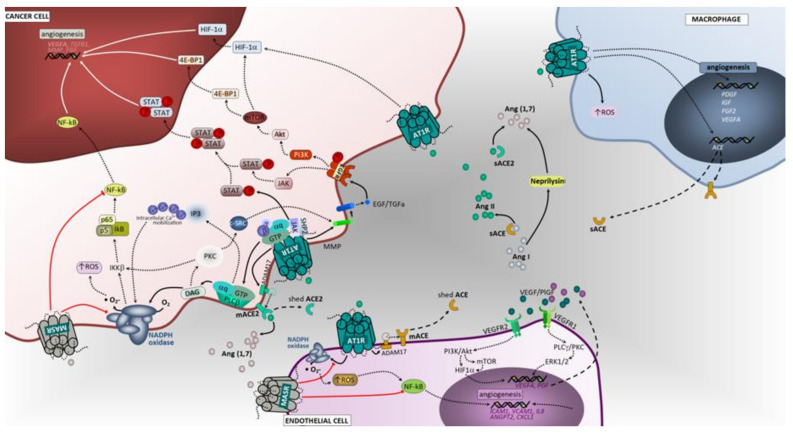
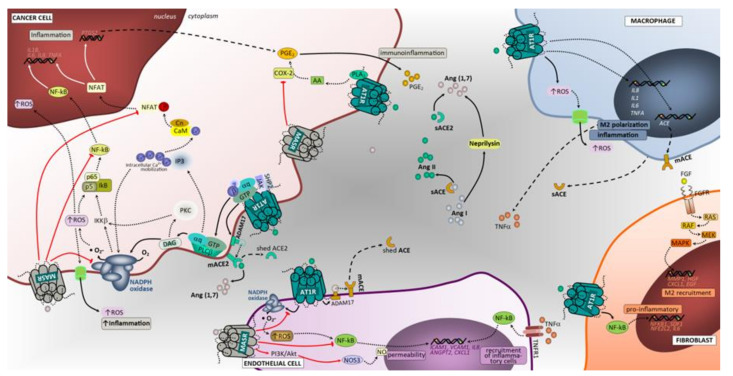
Figure 2
Angiotensin-associated pathways associated with cell proliferation, invasion, and migration in lung tumor microenvironment. Membrane-bound and soluble ACE and ACE2 catalyze the production of angiotensin II or angiotensin (1,7), ligands that exert regulatory functions in tumor microenvironment cells. The effects of activating Ang II/AT1R, Ang II/AT2R and Ang (1,7)/MasR axes’ signaling has been mostly studied in tumor cells, including lung cancer cells. Overall, in contrast to the Ang II/AT1R that mediates several pathological events associated with activated RAS, the Ang (1,7)/MasR and Ang II/AT2R pathways are thought to antagonize many of the cellular actions of the Ang II-AT1R axis. In cancer cells, upon binding of Ang II to AT1R, a pleiotropic downstream signaling cascade is triggered, ultimately causing, either directly or indirectly, upregulation of cell proliferation, survival, motility, migration, invasion and EMT. Activated AT1R subunits stimulate PLCβ, that hydrolyses membrane lipids, activates PKC and mobilization of intracellular Ca2+, while free Gβ and Gγ subunits bind and gate ion channels. Activated Gαq/11 units also activate the JAK-SHP2/STAT pathway and receptor tyrosine kinase (RTK) transactivation. Activation of RTK, depicted in the figure as EGFR, occurs through second messenger’s stimulation of ADAM family and MMPs to cleave its ligands (in the figure EGF and TGFα) that bind and activate RTK. Alternatively, the AT1R-mediated activation of MMPs can follow a PLCβ/DAG/PKC/c-SRC signaling mechanism to elicit increased ligands for RTK. Subsequent signaling upon activation of EGFR is represented in the figure using dashed arrows. Other intracellular cascades mediated by Ang II/AT1R include the activation of CXCR4/SDF-1 signaling through FAK/RhoA/ROCK1-2/MLC increasing cell contraction, migratory potential and tumor invasion. The Ang (1,7)/MasR activation inhibits NFAT transcriptional regulation that reduces proliferation. Notably, this pathway blocks the NF-kB molecule formation thereby impacting EMT (including Snail1-mediated), invasion and survival. In addition, the inhibitory effect over ERK1/2 and NAPH oxidase signaling pathways significantly impact cell proliferation. The Ang II/AT2R pathway signals are mediated through protein phosphatases PTP1B, PTP and PP2A. The inhibition of CAV-1 phosphorylation stops the Rab5/Rac1/GTP migratory potential of malignant cells, thus suppressing invasion. Furthermore, AT2R-associated increase in PTP and PP2A exerts blocking effects in RTK-mediated signals of the RAS/RAF/MEK1-2/ERK1-2 pathway at the level of MERK1/2 and RAS molecules, reducing cell proliferation. Macrophages, endothelial cells and fibroblasts are important components of the tumor microenvironment and capable of generating and expressing RAS components. These cells, beyond their functional ligands and receptors that are altered in tumor microenvironment, and reflect the crosstalk between all cell constituents, also use the RAS signaling pathway (mostly AT1R, but also MasR in endothelial cells) to yield functional characteristics that ultimately may favor the cell itself and enhance tumor growth. BIRC5, surviving gene; BCL2, B-cell leukemia/lymphoma 2; BAD, BCL2 associated agonist of cell death; GADD45, Growth Arrest and DNA Damage 45; SREBP, Sterol regulatory element binding proteins; ROCK1/2, Rho-associated protein kinase 1/2; Rab5, Ras-related protein Rab-5; S6K, ribosomal S6 kinase; PTP, protein tyrosine phosphatase; PP2A, Protein phosphatase 2; 4E-BP1, Eukaryotic translation initiation factor 4E-binding protein 1; IKKB, Inhibitor of nuclear factor kappa-B kinase subunit beta; c-SRC, cellular Proto-oncogene tyrosine-protein kinase; SHP2, Src homology region 2 domain-containing phosphatase-2; PTP1B, Protein tyrosine phosphatase 1B; MLC, myosin light chain; CAV1, caveolin 1; IL1, interleukin 1; TNFA, tumor necrosis factor; Snail1, Zinc finger protein SNAI1; SNAI1, Snail Family Transcriptional Repressor 1; JAK, janus kinase; STAT, signal transduction and activator of transcription; GTP, guanosine triphosphate; EGF, epidermal growth factor; EGFR, epidermal growth factor receptor; CXCL1, The chemokine (C-X-C motif) ligand 1; IL8, interleukin 8; NF-kB, Nuclear Factor-kappa B; SDF1, stromal cell-derived factor 1; IL6, interleukin 6; HGF, hepatocyte growth factor; RAS, oncogene protein p21; FGF, fibroblast growth factor; FGFR, fibroblast growth factor receptor; Cas9, caspase 9; Cas3, caspase 3; ERK1/2, extracellular signal-regulated kinase; PI3K/Akt, phosphoinositide 3-kinase/protein kinase B; MEK1/2, mitogen activated protein kinase kinase; HIF1α, hypoxia inducible factor 1 alpha; NO, nitric oxide; RhoA, Ras homolog family member A; MAPK, mitogen activated protein kinase; mTOR, mammalian target of rapamycin; IP3, inositol trisphosphate; DAG, diacylglycerol; M2, macrophage polarized towards M2; ADAM17, Desintegrin and metalloproteinase domain-containing protein 17; p38/MAPK, protein 38/mitogen activated protein kinase; AT1R, angiotensin receptor 1; AT2R, angiotensin receptor 2; MasR, G-protein coupled Mas receptor; VEGF, vascular endothelial growth factor; NADPH, reduced form of nicotinamide adenine dinucleotide phosphate; mACE, membrane angiotensin converting enzyme; sACE, soluble angiotensin converting enzyme; mACE2, membrane angiotensin converting enzyme 2; sACE2, soluble angiotensin converting enzyme 2; TGFBR, transforming growth factor beta receptor; VEGFR2, vascular endothelial growth factor receptor 2; TGFα, transforming growth factor alpha; Ang (1,7), angiotensin 1,7; PLCβ, phospholipase C beta; FAK, Focal adhesion kinase; NFAT, nuclear factor of activated T cells.