

Linker histone epitopes are hidden by in situ higher-order chromatin structure
- PMID: 32505195
- PMCID: PMC7276084
- DOI: 10.1186/s13072-020-00345-9
Linker histone epitopes are hidden by in situ higher-order chromatin structure
Abstract
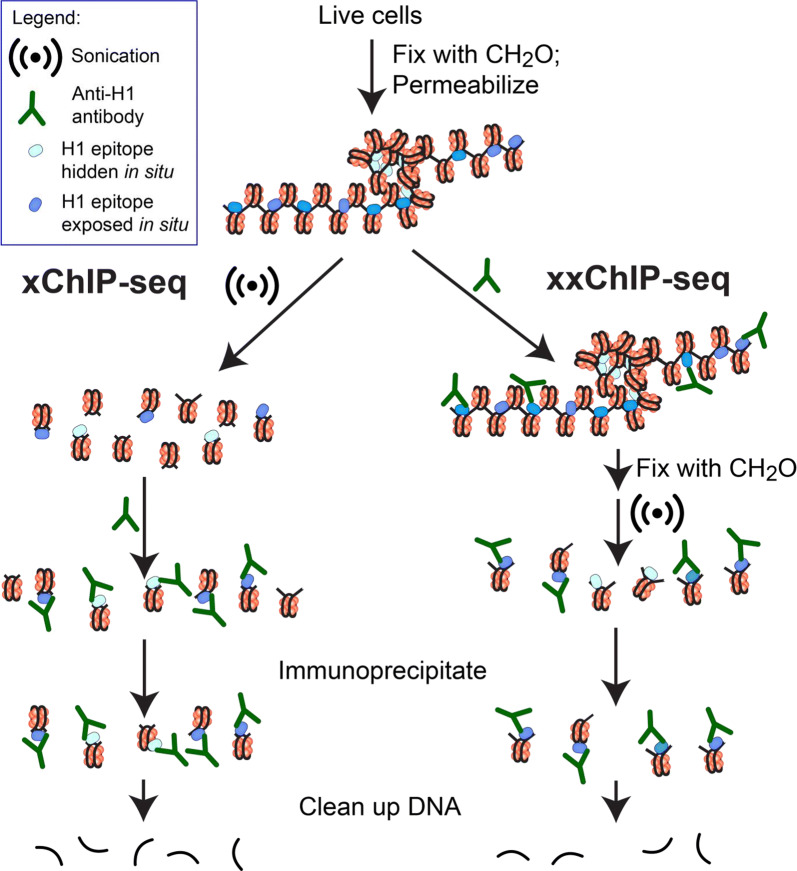
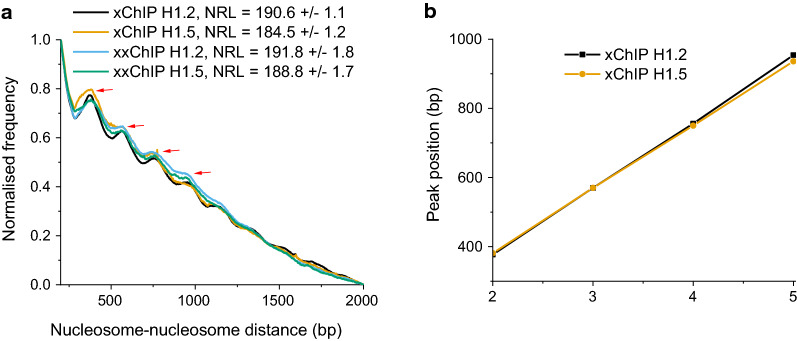
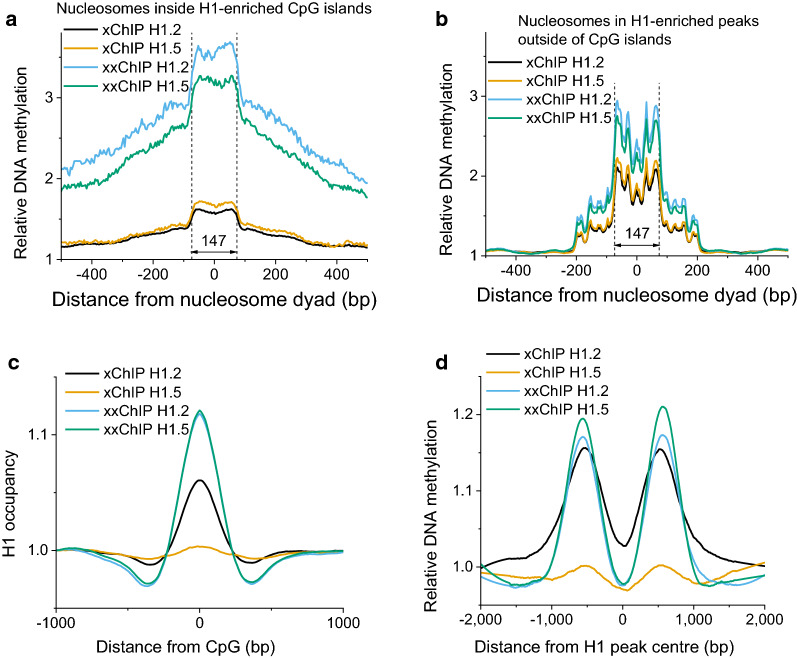
Background: Histone H1 is the most mobile histone in the cell nucleus. Defining the positions of H1 on chromatin in situ, therefore, represents a challenge. Immunoprecipitation of formaldehyde-fixed and sonicated chromatin, followed by DNA sequencing (xChIP-seq), is traditionally the method for mapping histones onto DNA elements. But since sonication fragmentation precedes ChIP, there is a consequent loss of information about chromatin higher-order structure. Here, we present a new method, xxChIP-seq, employing antibody binding to fixed intact in situ chromatin, followed by extensive washing, a second fixation, sonication and immunoprecipitation. The second fixation is intended to prevent the loss of specifically bound antibody during washing and subsequent sonication and to prevent antibody shifting to epitopes revealed by the sonication process. In many respects, xxChIP-seq is comparable to immunostaining microscopy, which also involves interaction of the primary antibody with fixed and permeabilized intact cells. The only epitopes displayed after immunostaining are the "exposed" epitopes, not "hidden" by the fixation of chromatin higher-order structure. Comparison of immunoprecipitated fragments between xChIP-seq versus xxChIP-seq should indicate which epitopes become inaccessible with fixation and identify their associated DNA elements.
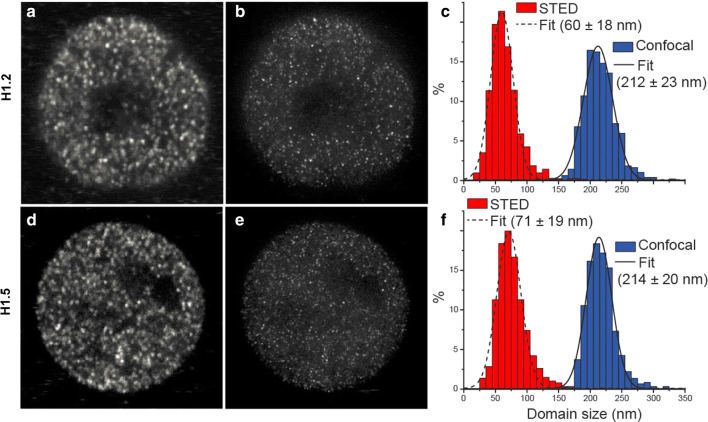
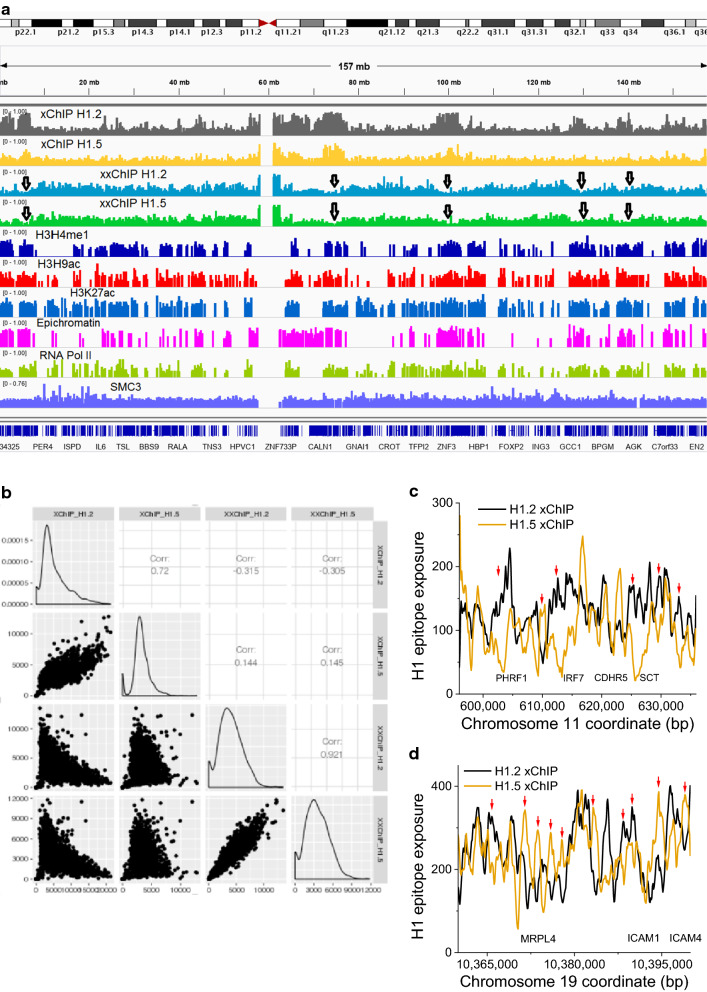
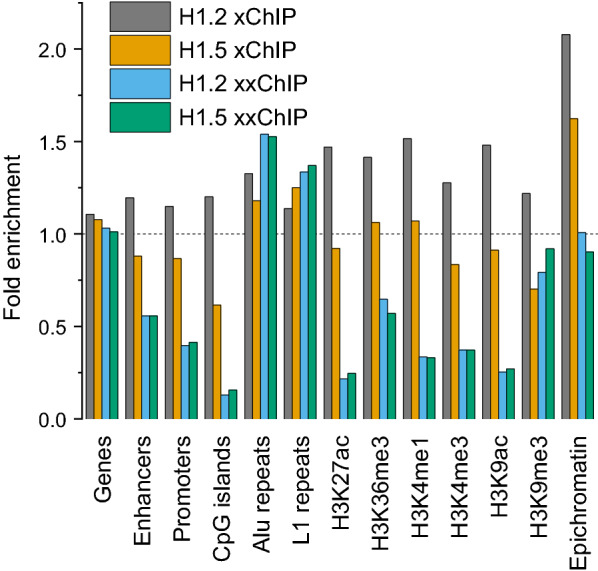
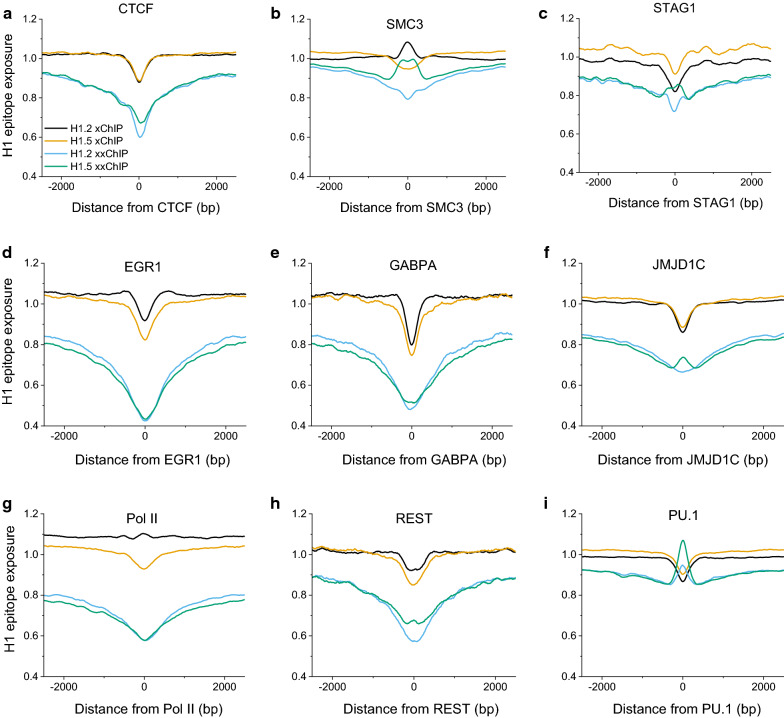
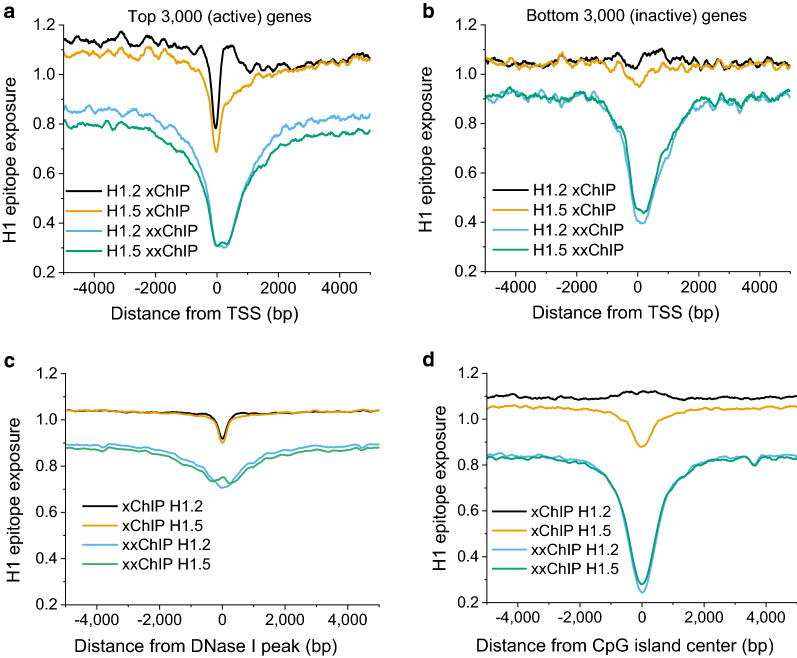
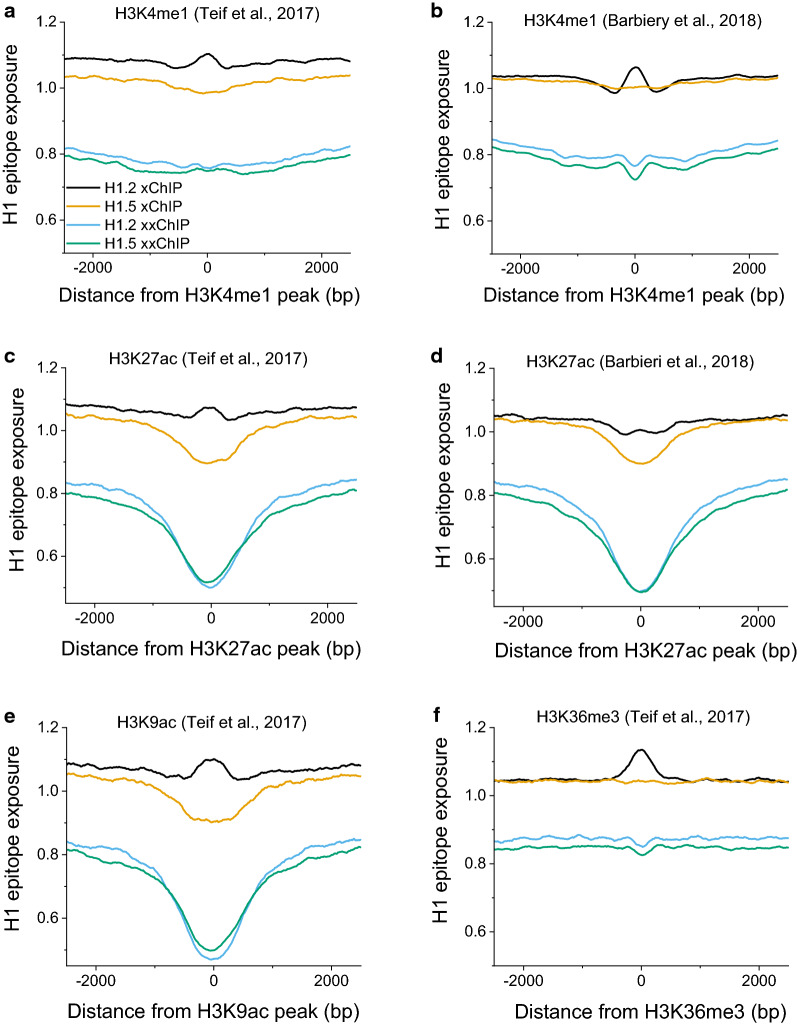
Results: We determined the genomic distribution of histone variants H1.2 and H1.5 in human myeloid leukemia cells HL-60/S4 and compared their epitope exposure by both xChIP-seq and xxChIP-seq, as well as high-resolution microscopy, illustrating the influences of preserved chromatin higher-order structure in situ. We found that xChIP and xxChIP H1 signals are in general negatively correlated, with differences being more pronounced near active regulatory regions. Among the intriguing observations, we find that transcription-related regions and histone PTMs (i.e., enhancers, promoters, CpG islands, H3K4me1, H3K4me3, H3K9ac, H3K27ac and H3K36me3) exhibit significant deficiencies (depletions) in H1.2 and H1.5 xxChIP-seq reads, compared to xChIP-seq. These observations suggest the existence of in situ transcription-related chromatin higher-order structures stabilized by formaldehyde.
Conclusion: Comparison of H1 xxChIP-seq to H1 xChIP-seq allows the development of hypotheses on the chromosomal localization of (stabilized) higher-order structure, indicated by the generation of "hidden" H1 epitopes following formaldehyde crosslinking. Changes in H1 epitope exposure surrounding averaged chromosomal binding sites or epigenetic modifications can also indicate whether these sites have chromatin higher-order structure. For example, comparison between averaged active or inactive promoter regions suggests that both regions can acquire stabilized higher-order structure with hidden H1 epitopes. However, the H1 xChIP-seq comparison cannot define their differences. Application of the xxChIP-seq versus H1 xChIP-seq method is particularly relevant to chromatin-associated proteins, such as linker histones, that play dynamic roles in establishing chromatin higher-order structure.
Conflict of interest statement
The authors declare that they have no competing interests.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Miscellaneous
