

Practically useful protein-design methods combining phylogenetic and atomistic calculations
- PMID: 32505941
- PMCID: PMC7289631
- DOI: 10.1016/j.sbi.2020.04.003
Practically useful protein-design methods combining phylogenetic and atomistic calculations
Abstract
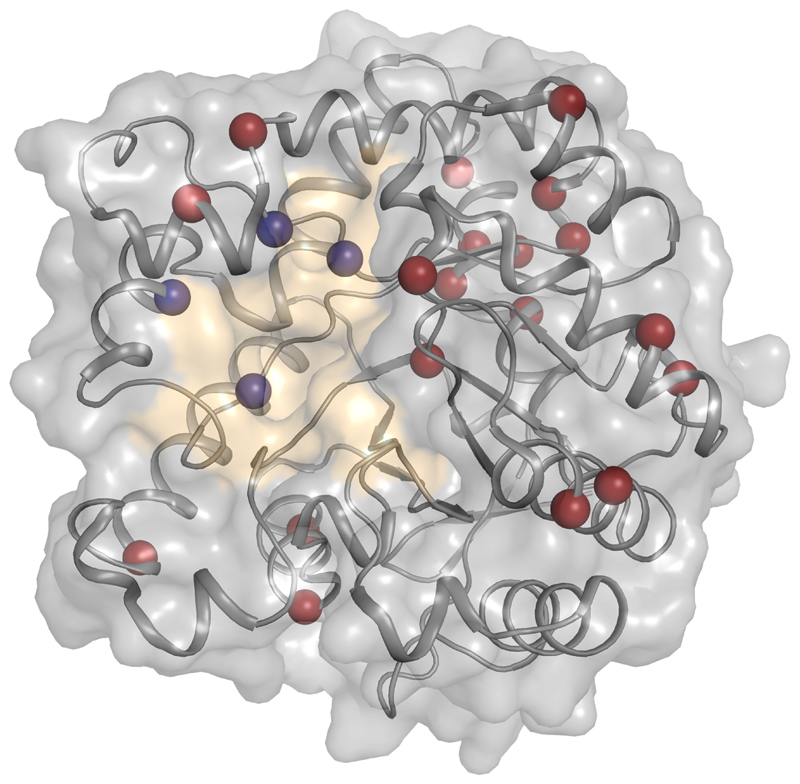
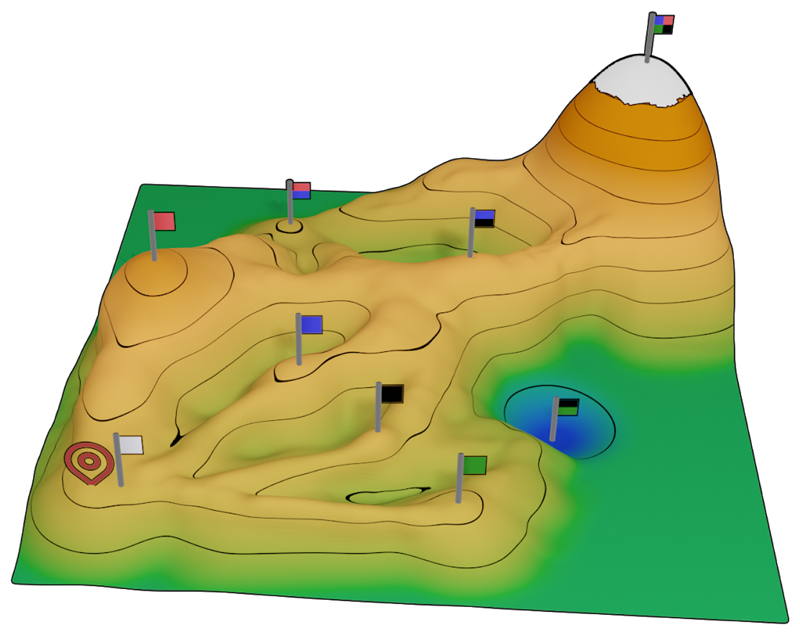
Our ability to design new or improved biomolecular activities depends on understanding the sequence-function relationships in proteins. The large size and fold complexity of most proteins, however, obscure these relationships, and protein-optimization methods continue to rely on laborious experimental iterations. Recently, a deeper understanding of the roles of stability-threshold effects and biomolecular epistasis in proteins has led to the development of hybrid methods that combine phylogenetic analysis with atomistic design calculations. These methods enable reliable and even single-step optimization of protein stability, expressibility, and activity in proteins that were considered outside the scope of computational design. Furthermore, ancestral-sequence reconstruction produces insights on missing links in the evolution of enzymes and binders that may be used in protein design. Through the combination of phylogenetic and atomistic calculations, the long-standing goal of general computational methods that can be universally applied to study and optimize proteins finally seems within reach.
Copyright © 2020 Elsevier Ltd. All rights reserved.
Figures

References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
