

Nitroxyl: A Novel Strategy to Circumvent Diabetes Associated Impairments in Nitric Oxide Signaling
- PMID: 32508651
- PMCID: PMC7248192
- DOI: 10.3389/fphar.2020.00727
Nitroxyl: A Novel Strategy to Circumvent Diabetes Associated Impairments in Nitric Oxide Signaling
Abstract
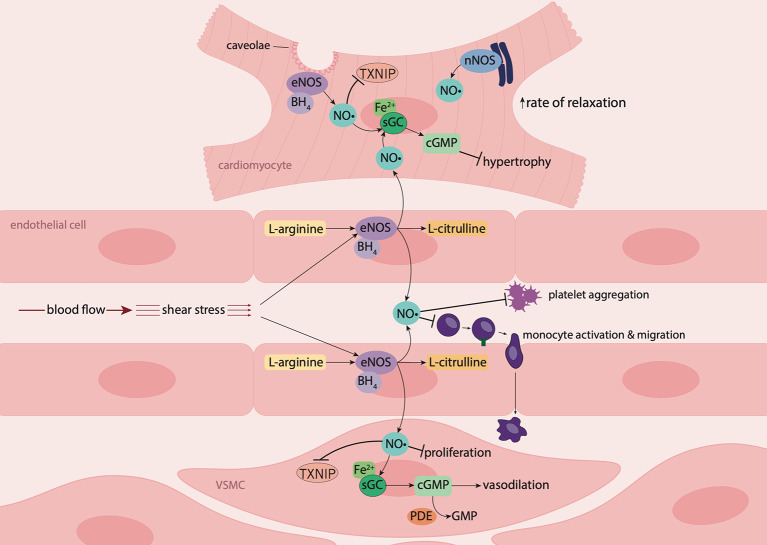
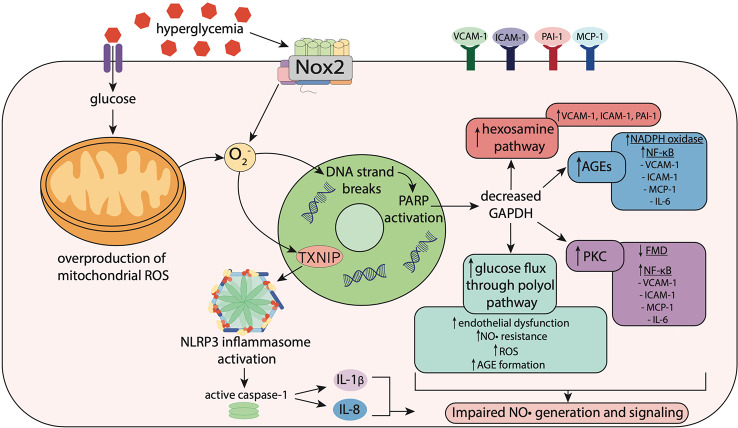
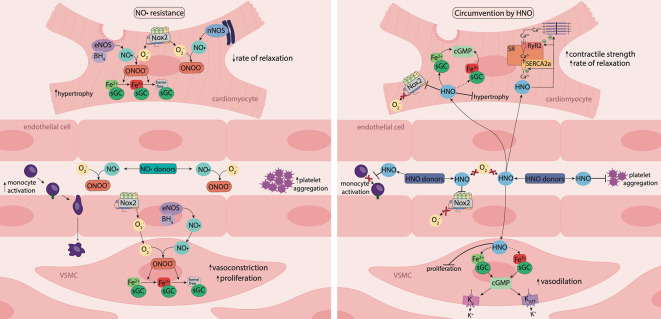
Diabetes is associated with an increased mortality risk due to cardiovascular complications. Hyperglycemia-induced oxidative stress underlies these complications, leading to an impairment in endogenous nitric oxide (NO•) generation, together with reductions in NO• bioavailability and NO• responsiveness in the vasculature, platelets and myocardium. The latter impairment of responsiveness to NO•, termed NO• resistance, compromises the ability of traditional NO•-based therapeutics to improve hemodynamic status during diabetes-associated cardiovascular emergencies, such as acute myocardial infarction. Whilst a number of agents can ameliorate (e.g. angiotensin converting enzyme [ACE] inhibitors, perhexiline, statins and insulin) or circumvent (e.g. nitrite and sGC activators) NO• resistance, nitroxyl (HNO) donors offer a novel opportunity to circumvent NO• resistance in diabetes. With a suite of vasoprotective properties and an ability to enhance cardiac inotropic and lusitropic responses, coupled with preserved efficacy in the setting of oxidative stress, HNO donors have intact therapeutic potential in the face of diminished NO• signaling. This review explores the major mechanisms by which hyperglycemia-induced oxidative stress drives NO• resistance, and the therapeutic potential of HNO donors to circumvent this to treat cardiovascular complications in type 2 diabetes mellitus.
Keywords: HNO; cardiovascular disease; diabetes; nitric oxide; nitric oxide resistance; nitroxyl; type 2 diabetes.
Copyright © 2020 Velagic, Qin, Woodman, Horowitz, Ritchie and Kemp-Harper.
Figures
References
-
- Andrews K. L., Irvine J. C., Tare M., Apostolopoulos J., Favaloro J. L., Triggle C. R., et al. (2009). A role for nitroxyl (HNO) as an endothelium-derived relaxing and hyperpolarizing factor in resistance arteries. Br. J. Pharmacol. 157, 540–550. 10.1111/j.1476-5381.2009.00150.x - DOI - PMC - PubMed
Publication types
LinkOut - more resources
Full Text Sources
Miscellaneous