

How Do You Identify m6 A Methylation in Transcriptomes at High Resolution? A Comparison of Recent Datasets
- PMID: 32508872
- PMCID: PMC7251061
- DOI: 10.3389/fgene.2020.00398
How Do You Identify m6 A Methylation in Transcriptomes at High Resolution? A Comparison of Recent Datasets
Abstract
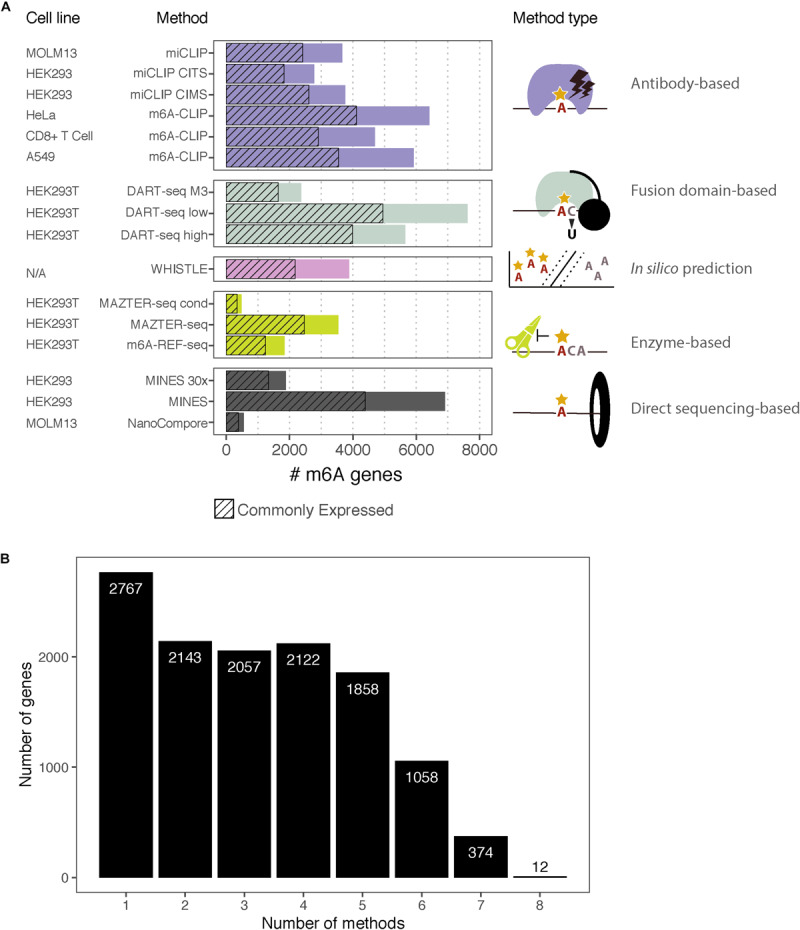
A flurry of methods has been developed in recent years to identify N6-methyladenosine (m6A) sites across transcriptomes at high resolution. This raises the need to understand both the common features and those that are unique to each method. Here, we complement the analyses presented in the original papers by reviewing their various technical aspects and comparing the overlap between m6A-methylated messenger RNAs (mRNAs) identified by each. Specifically, we examine eight different methods that identify m6A sites in human cells with high resolution: two antibody-based crosslinking and immunoprecipitation (CLIP) approaches, two using endoribonuclease MazF, one based on deamination, two using Nanopore direct RNA sequencing, and finally, one based on computational predictions. We contrast the respective datasets and discuss the challenges in interpreting the overlap between them, including a prominent expression bias in detected genes. This overview will help guide researchers in making informed choices about using the available data and assist with the design of future experiments to expand our understanding of m6A and its regulation.
Keywords: N6-methyladenosine; RNA; bioinformatics; epitranscriptomics; m6A.
Copyright © 2020 Capitanchik, Toolan-Kerr, Luscombe and Ule.
Figures


References
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources