

TOOme: A Novel Computational Framework to Infer Cancer Tissue-of-Origin by Integrating Both Gene Mutation and Expression
- PMID: 32509741
- PMCID: PMC7248358
- DOI: 10.3389/fbioe.2020.00394
TOOme: A Novel Computational Framework to Infer Cancer Tissue-of-Origin by Integrating Both Gene Mutation and Expression
Abstract
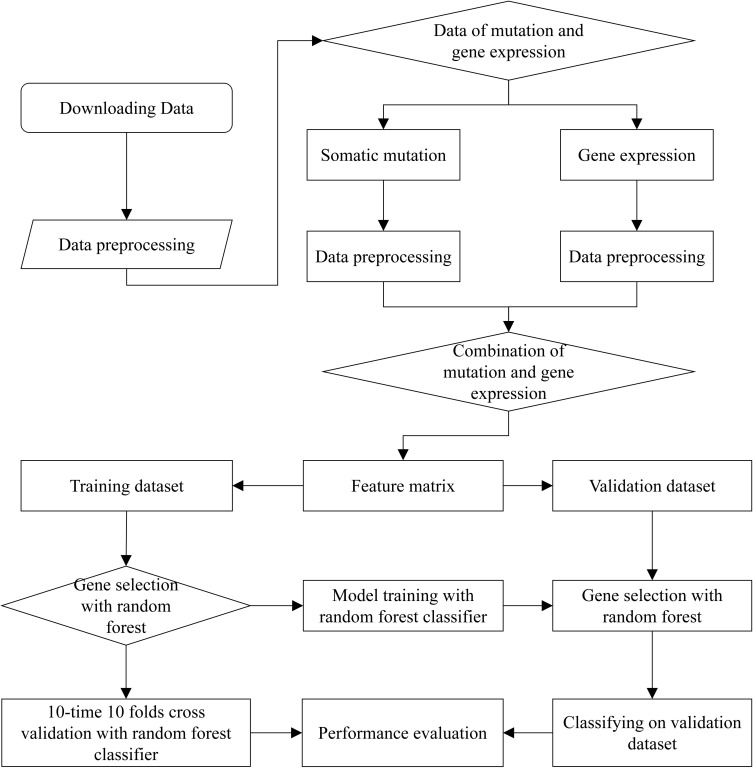
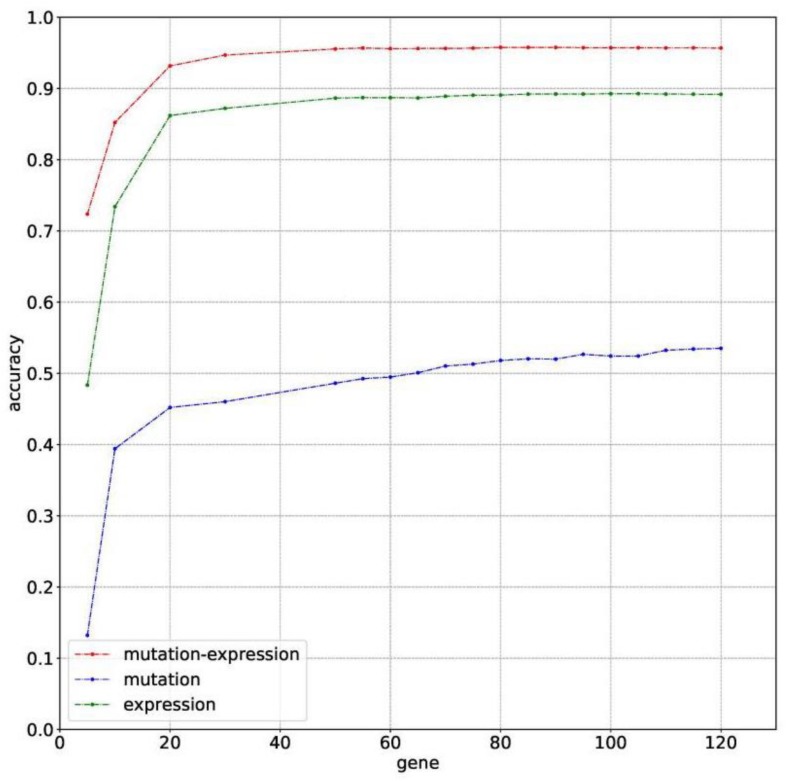
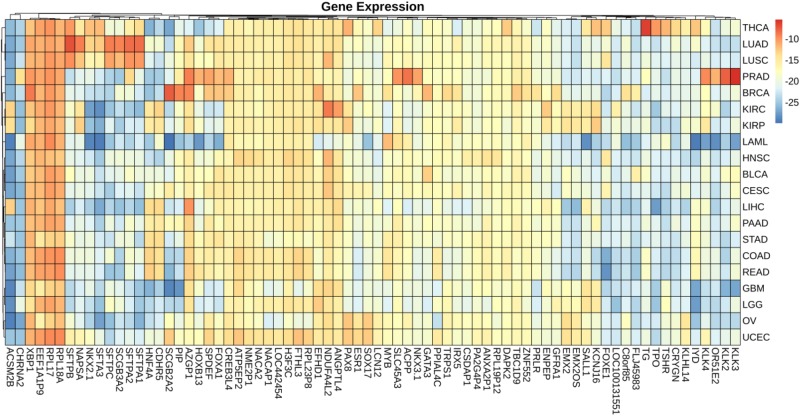
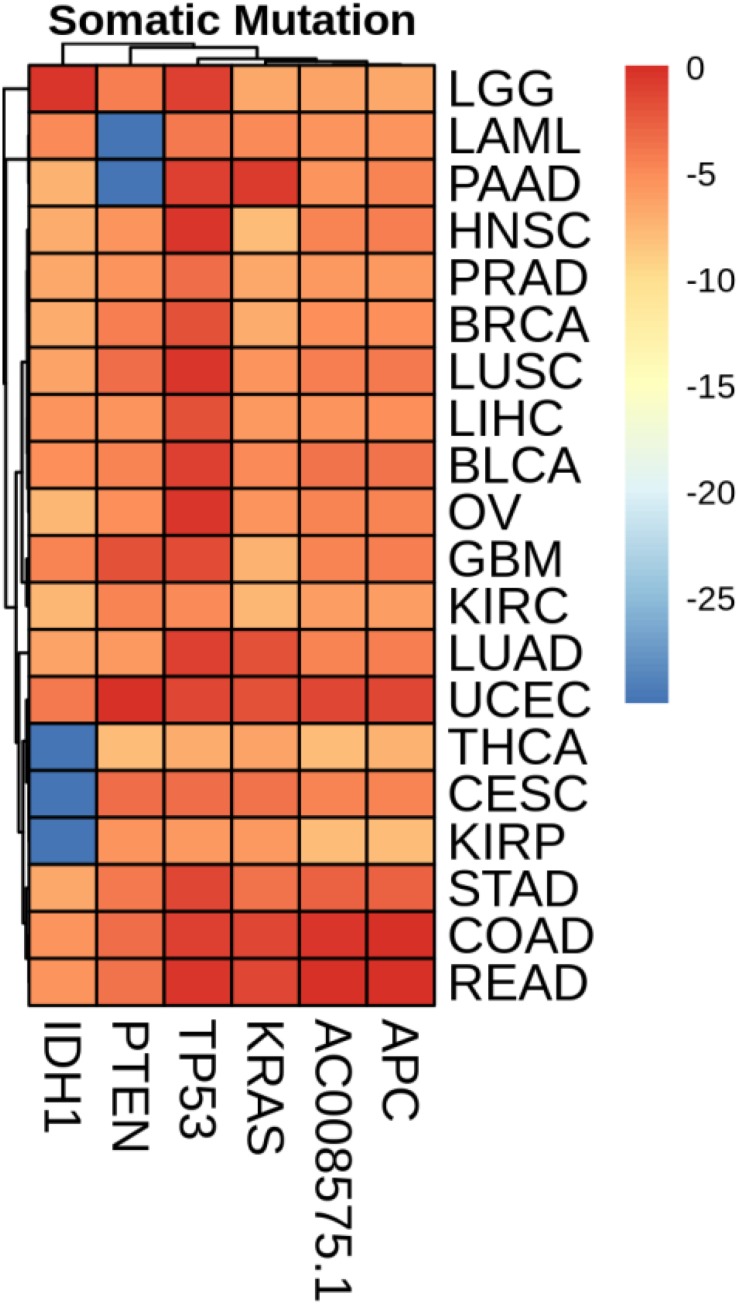
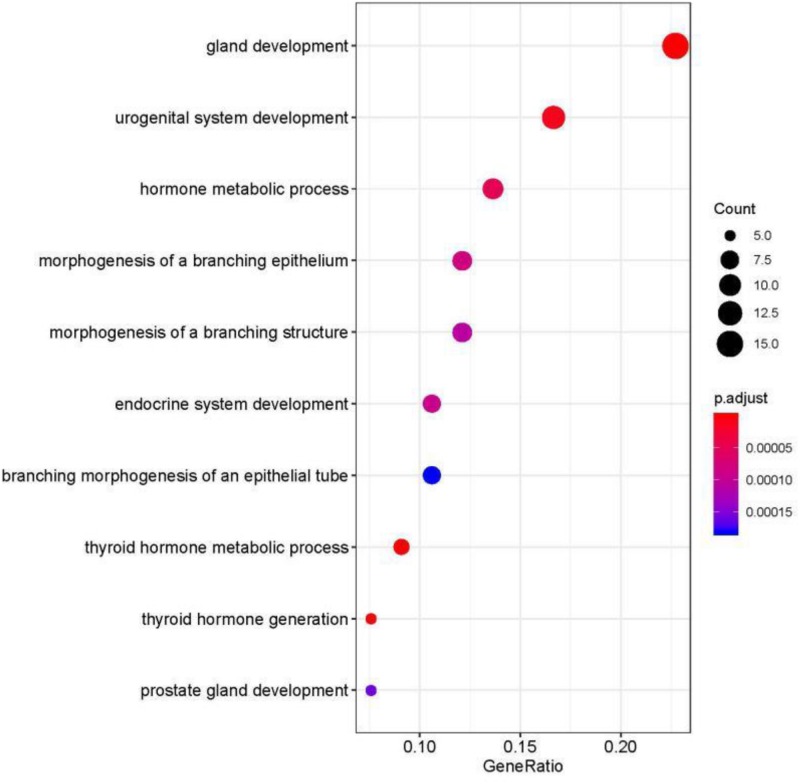
Metastatic cancers require further diagnosis to determine their primary tumor sites. However, the tissue-of-origin for around 5% tumors could not be identified by routine medical diagnosis according to a statistics in the United States. With the development of machine learning techniques and the accumulation of big cancer data from The Cancer Genome Atlas (TCGA) and Gene Expression Omnibus (GEO), it is now feasible to predict cancer tissue-of-origin by computational tools. Metastatic tumor inherits characteristics from its tissue-of-origin, and both gene expression profile and somatic mutation have tissue specificity. Thus, we developed a computational framework to infer tumor tissue-of-origin by integrating both gene mutation and expression (TOOme). Specifically, we first perform feature selection on both gene expressions and mutations by a random forest method. The selected features are then used to build up a multi-label classification model to infer cancer tissue-of-origin. We adopt a few popular multiple-label classification methods, which are compared by the 10-fold cross validation process. We applied TOOme to the TCGA data containing 7,008 non-metastatic samples across 20 solid tumors. Seventy four genes by gene expression profile and six genes by gene mutation are selected by the random forest process, which can be divided into two categories: (1) cancer type specific genes and (2) those expressed or mutated in several cancers with different levels of expression or mutation rates. Function analysis indicates that the selected genes are significantly enriched in gland development, urogenital system development, hormone metabolic process, thyroid hormone generation prostate hormone generation and so on. According to the multiple-label classification method, random forest performs the best with a 10-fold cross-validation prediction accuracy of 96%. We also use the 19 metastatic samples from TCGA and 256 cancer samples downloaded from GEO as independent testing data, for which TOOme achieves a prediction accuracy of 89%. The cross-validation validation accuracy is better than those using gene expression (i.e., 95%) and gene mutation (53%) alone. In conclusion, TOOme provides a quick yet accurate alternative to traditional medical methods in inferring cancer tissue-of-origin. In addition, the methods combining somatic mutation and gene expressions outperform those using gene expression or mutation alone.
Keywords: cross-validation; gene expression; random forest; somatic mutation; tissue-of-origin.
Copyright © 2020 He, Lang, Wang, Liu, Lu, He, Gao, Bing, Tian and Yang.
Figures
References
LinkOut - more resources
Full Text Sources