

This is a preprint.
Fast Identification of Possible Drug Treatment of Coronavirus Disease -19 (COVID-19) Through Computational Drug Repurposing Study
- PMID: 32510523
- PMCID: PMC7263765
- DOI: 10.26434/chemrxiv.11875446
Fast Identification of Possible Drug Treatment of Coronavirus Disease -19 (COVID-19) Through Computational Drug Repurposing Study
Update in
-
Fast Identification of Possible Drug Treatment of Coronavirus Disease-19 (COVID-19) through Computational Drug Repurposing Study.J Chem Inf Model. 2020 Jun 22;60(6):3277-3286. doi: 10.1021/acs.jcim.0c00179. Epub 2020 May 4. J Chem Inf Model. 2020. PMID: 32315171 Free PMC article.
Abstract
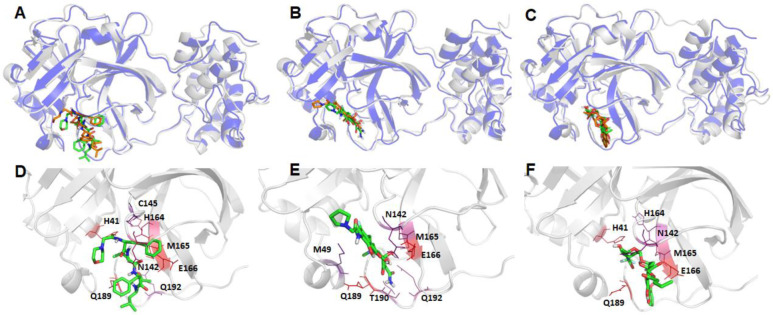
The recent outbreak of novel coronavirus disease -19 (COVID-19) calls for and welcomes possible treatment strategies using drugs on the market. It is very efficient to apply computer-aided drug design techniques to quickly identify promising drug repurposing candidates, especially after the detailed 3D-structures of key virous proteins are resolved. Taking the advantage of a recently released crystal structure of COVID-19 protease in complex with a covalently-bonded inhibitor, N3,1 I conducted virtual docking screening of approved drugs and drug candidates in clinical trials. For the top docking hits, I then performed molecular dynamics simulations followed by binding free energy calculations using an endpoint method called MM-PBSA-WSAS.2-4 Several promising known drugs stand out as potential inhibitors of COVID-19 protease, including Carfilzomib, Eravacycline, Valrubicin, Lopinavir and Elbasvir. Carfilzomib, an approved anti-cancer drug acting as a proteasome inhibitor, has the best MM-PBSA-WSAS binding free energy, -13.82 kcal/mol. Streptomycin, an antibiotic and a charged molecule, also demonstrates some inhibitory effect, even though the predicted binding free energy of the charged form (-3.82 kcal/mol) is not nearly as low as that of the neutral form (-7.92 kcal/mol). One bioactive, PubChem 23727975, has a binding free energy of -12.86 kcal/mol. Detailed receptor-ligand interactions were analyzed and hot spots for the receptor-ligand binding were identified. I found that one hotspot residue HIS41, is a conserved residue across many viruses including COVID-19, SARS, MERS, and HCV. The findings of this study can facilitate rational drug design targeting the COVID-19 protease.
Conflict of interest statement
The author declares no competing financial interest.
Figures








References
-
- Liu X.; Zhang B.; Jin Z.; Yang H.; Rao Z., The crystal structure of COVID-19 main protease in complex with an inhibitor N3. 2020.
-
- Wang E.; Sun H.; Wang J.; Wang Z.; Liu H.; Zhang J. Z. H.; Hou T., End-Point Binding Free Energy Calculation with MM/PBSA and MM/GBSA: Strategies and Applications in Drug Design. Chem Rev 2019, 119 (16), 9478–9508. - PubMed
-
- Wang J. M.; Hou T. J.; Xu X. J., Recent Advances in Free Energy Calculations with a Combination of Molecular Mechanics and Continuum Models. Curr Comput-Aid Drug 2006, 2 (3), 287–306.
-
- Law V.; Knox C.; Djoumbou Y.; Jewison T.; Guo A. C.; Liu Y.; Maciejewski A.; Arndt D.; Wilson M.; Neveu V.; Tang A.; Gabriel G.; Ly C.; Adamjee S.; Dame Z. T.; Han B.; Zhou Y.; Wishart D. S., DrugBank 4.0: shedding new light on drug metabolism. Nucleic acids research 2014, 42 (Database issue), D1091–7. - PMC - PubMed
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous