

Within-Host Viral Diversity: A Window into Viral Evolution
- PMID: 32511081
- PMCID: PMC10150642
- DOI: 10.1146/annurev-virology-010320-061642
Within-Host Viral Diversity: A Window into Viral Evolution
Abstract
The evolutionary dynamics of a virus can differ within hosts and across populations. Studies of within-host evolution provide an important link between experimental studies of virus evolution and large-scale phylodynamic analyses. They can determine the extent to which global processes are recapitulated on local scales and how accurately experimental infections model natural ones. They may also inform epidemiologic models of disease spread and reveal how host-level dynamics contribute to a virus's evolution at a larger scale. Over the last decade, advances in viral sequencing have enabled detailed studies of viral genetic diversity within hosts. I review how within-host diversity is sampled, measured, and expressed, and how comparative studies of viral diversity can be leveraged to elucidate a virus's evolutionary dynamics. These concepts are illustrated with detailed reviews of recent research on the within-host evolution of influenza virus, dengue virus, and cytomegalovirus.
Keywords: diversity; evolution; models; quasispecies; sequencing.
Figures
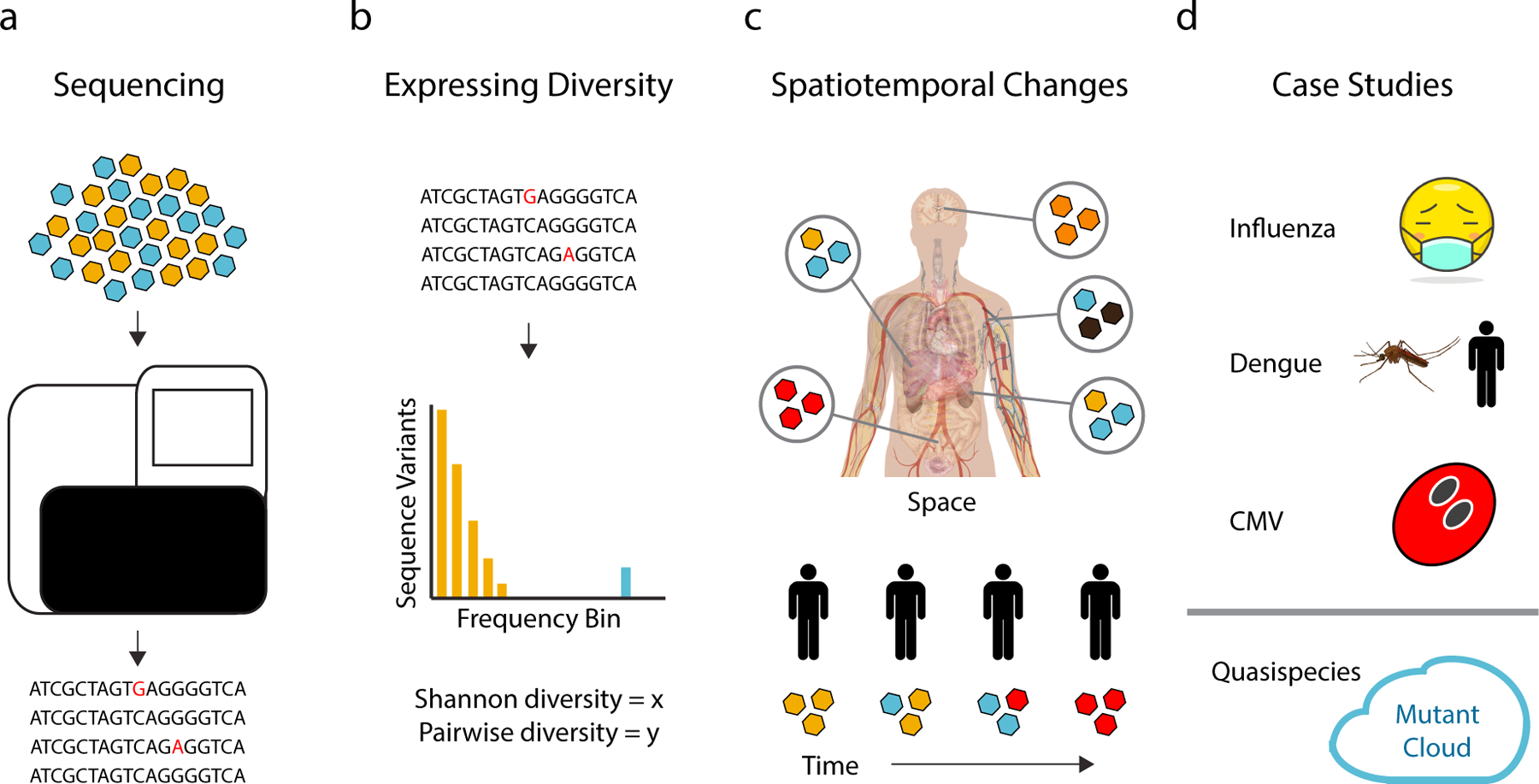
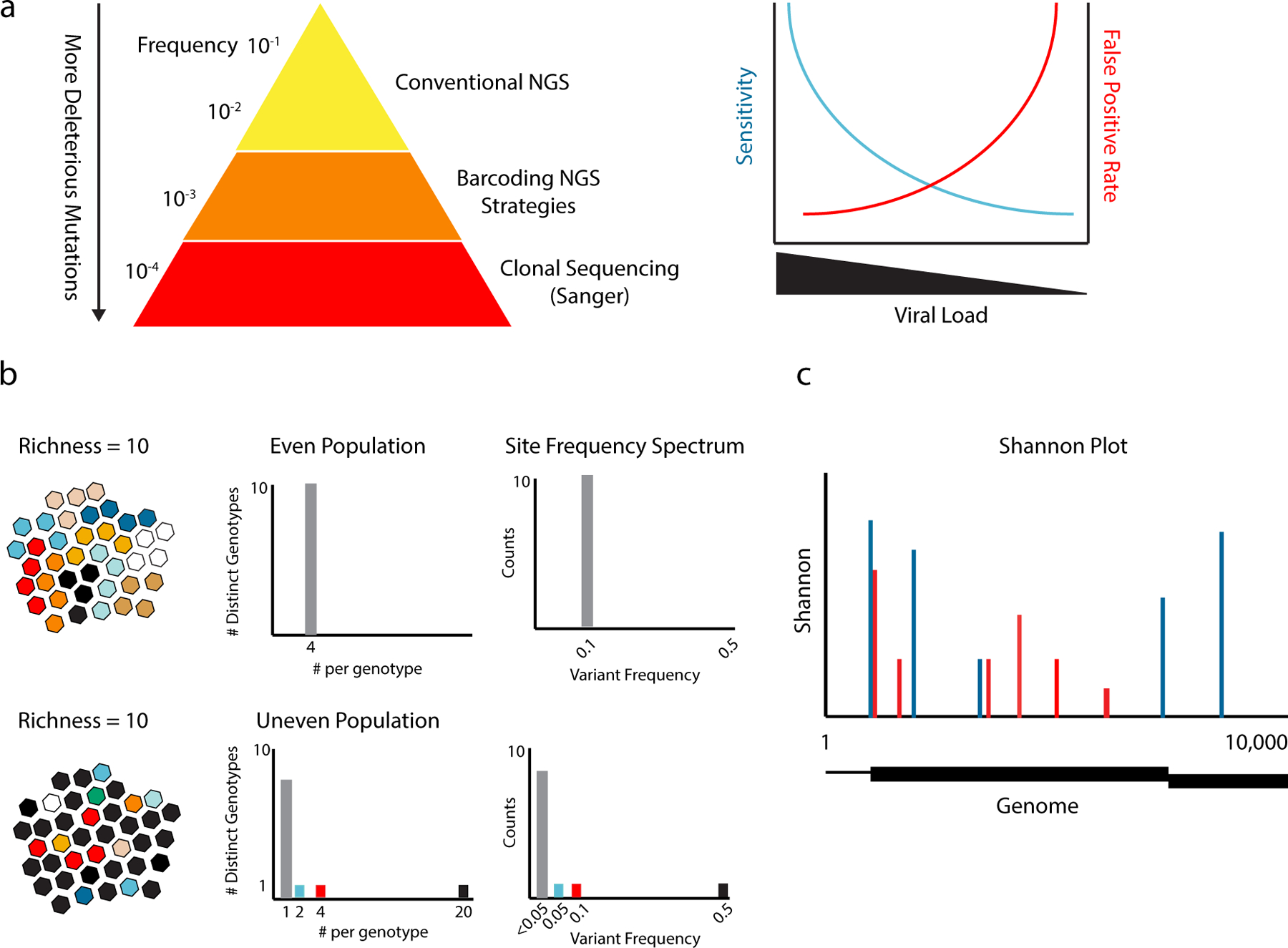
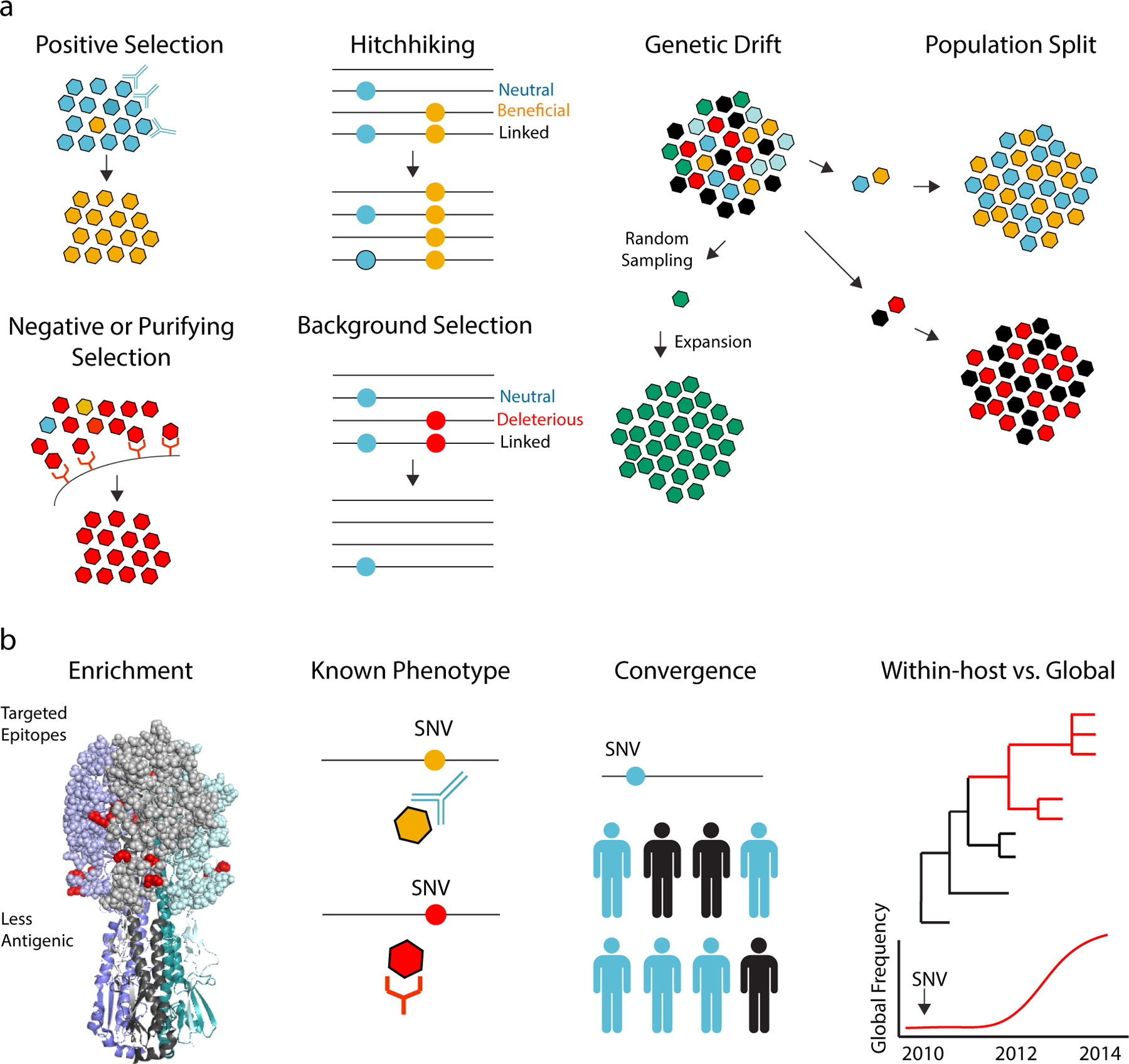
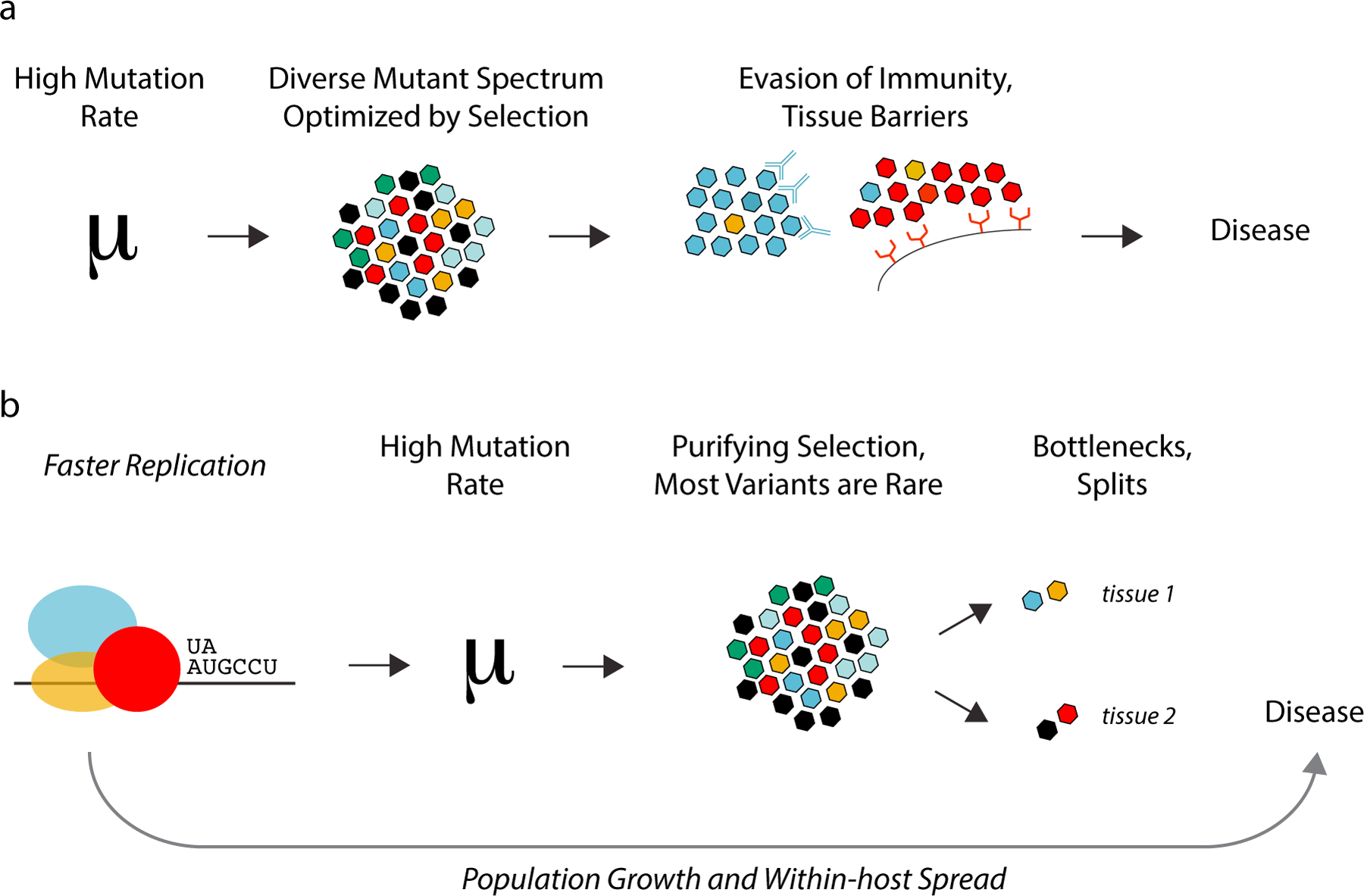
References
-
- Domingo E, Sabo D, Taniguchi T, Weissmann C. 1978. Nucleotide sequence heterogeneity of an RNA phage population. Cell 13:735–44 - PubMed
-
- Crotty S, Maag D, Arnold JJ, Zhong W, Lau JYN, et al. 2000. The broad-spectrum antiviral ribonucleoside ribavirin is an RNA virus mutagen. Nat. Med 6(12):1375–79 - PubMed
-
- Aaskov J. 2006. Long-Term Transmission of Defective RNA Viruses in Humans and Aedes Mosquitoes. Science 311(5758):236–38 - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
