

Evolution of Salmonella enterica serotype Typhimurium driven by anthropogenic selection and niche adaptation
- PMID: 32511244
- PMCID: PMC7302871
- DOI: 10.1371/journal.pgen.1008850
Evolution of Salmonella enterica serotype Typhimurium driven by anthropogenic selection and niche adaptation
Abstract
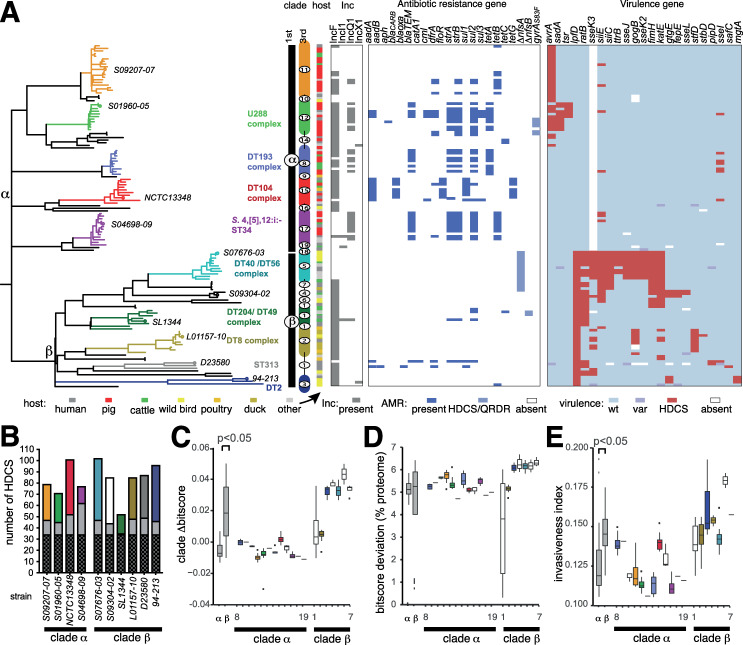
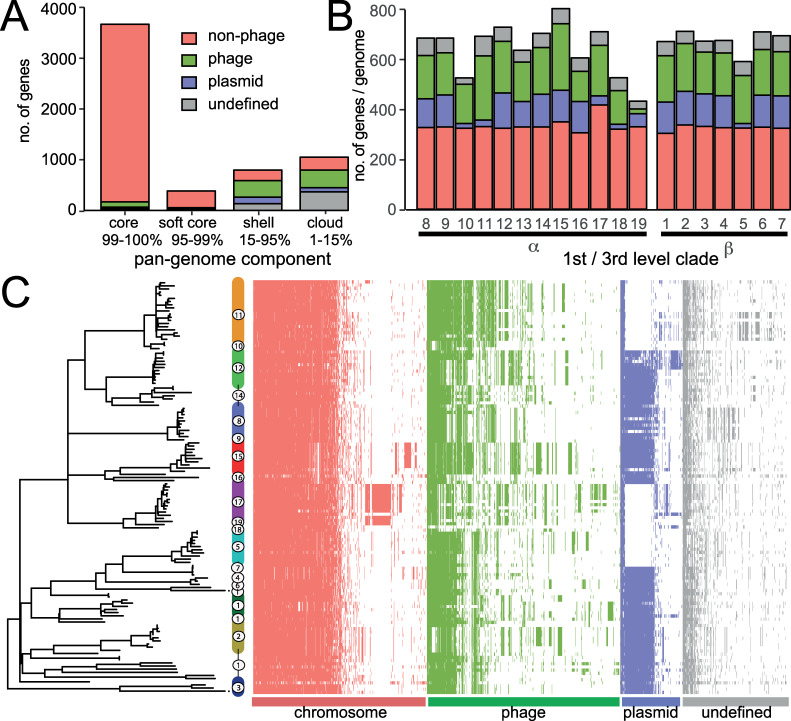
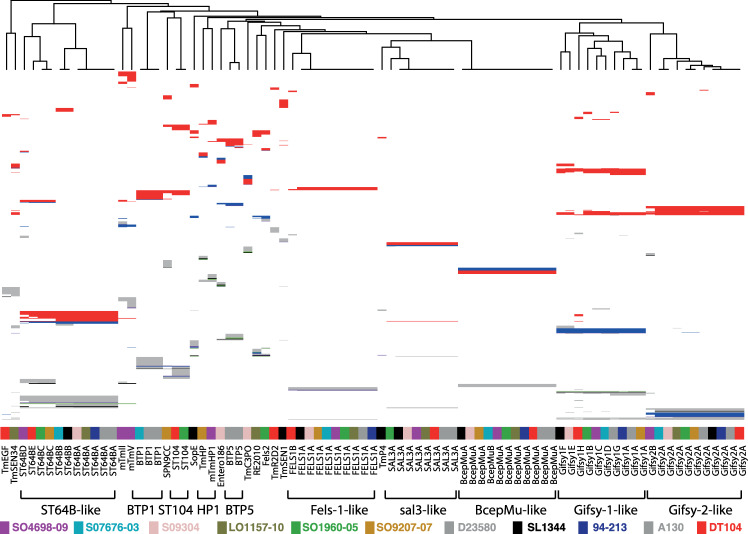
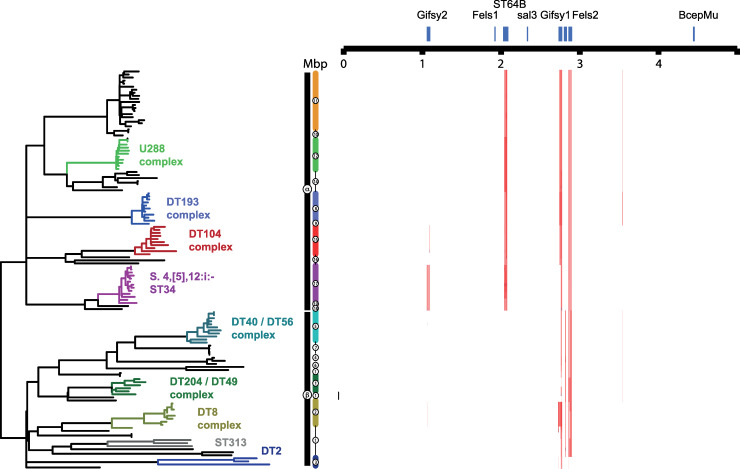
Salmonella enterica serotype Typhimurium (S. Typhimurium) is a leading cause of gastroenteritis and bacteraemia worldwide, and a model organism for the study of host-pathogen interactions. Two S. Typhimurium strains (SL1344 and ATCC14028) are widely used to study host-pathogen interactions, yet genotypic variation results in strains with diverse host range, pathogenicity and risk to food safety. The population structure of diverse strains of S. Typhimurium revealed a major phylogroup of predominantly sequence type 19 (ST19) and a minor phylogroup of ST36. The major phylogroup had a population structure with two high order clades (α and β) and multiple subclades on extended internal branches, that exhibited distinct signatures of host adaptation and anthropogenic selection. Clade α contained a number of subclades composed of strains from well characterized epidemics in domesticated animals, while clade β contained multiple subclades associated with wild avian species. The contrasting epidemiology of strains in clade α and β was reflected by the distinct distribution of antimicrobial resistance (AMR) genes, accumulation of hypothetically disrupted coding sequences (HDCS), and signatures of functional diversification. These observations were consistent with elevated anthropogenic selection of clade α lineages from adaptation to circulation in populations of domesticated livestock, and the predisposition of clade β lineages to undergo adaptation to an invasive lifestyle by a process of convergent evolution with of host adapted Salmonella serotypes. Gene flux was predominantly driven by acquisition and recombination of prophage and associated cargo genes, with only occasional loss of these elements. The acquisition of large chromosomally-encoded genetic islands was limited, but notably, a feature of two recent pandemic clones (DT104 and monophasic S. Typhimurium ST34) of clade α (SGI-1 and SGI-4).
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures

References
-
- Kirk MD, Pires SM, Black RE, Caipo M, Crump JA, Devleesschauwer B, et al. World Health Organization Estimates of the Global and Regional Disease Burden of 22 Foodborne Bacterial, Protozoal, and Viral Diseases, 2010: A Data Synthesis. PLoS Med. 2015;12(12):e1001921 Epub 2015/12/04. 10.1371/journal.pmed.1001921 - DOI - PMC - PubMed
-
- Anonymous. Salmonella in livestock production in Great Britain, 2017: gov.uk; 2018 [cited 2019 June 2019]. Available from: https://www.gov.uk/government/publications/salmonella-in-livestock-produ....
Publication types
MeSH terms
Grants and funding
- BB/M025489/1/BB_/Biotechnology and Biological Sciences Research Council/United Kingdom
- BB/J004529/1/BB_/Biotechnology and Biological Sciences Research Council/United Kingdom
- BBS/E/F/000PR10356/BB_/Biotechnology and Biological Sciences Research Council/United Kingdom
- BB/N007964/1/BB_/Biotechnology and Biological Sciences Research Council/United Kingdom
- BBS/E/F/000PR10353 /BB_/Biotechnology and Biological Sciences Research Council/United Kingdom
- BB/ CCG1860/1/BB_/Biotechnology and Biological Sciences Research Council/United Kingdom
- BB/R012490/1/BB_/Biotechnology and Biological Sciences Research Council/United Kingdom
- BBS/E/F/000PR10348 /BB_/Biotechnology and Biological Sciences Research Council/United Kingdom
- BBS/E/F/000PR10352/BB_/Biotechnology and Biological Sciences Research Council/United Kingdom
- BB/R012504/1/BB_/Biotechnology and Biological Sciences Research Council/United Kingdom
- BB/CCG1720/1/BB_/Biotechnology and Biological Sciences Research Council/United Kingdom
LinkOut - more resources
Full Text Sources
Medical
