

This is a preprint.
Recombination and lineage-specific mutations linked to the emergence of SARS-CoV-2
- PMID: 32511304
- PMCID: PMC7217262
- DOI: 10.1101/2020.02.10.942748
Recombination and lineage-specific mutations linked to the emergence of SARS-CoV-2
Update in
-
Recombination and lineage-specific mutations linked to the emergence of SARS-CoV-2.Genome Med. 2021 Aug 6;13(1):124. doi: 10.1186/s13073-021-00943-6. Genome Med. 2021. PMID: 34362430 Free PMC article.
Abstract
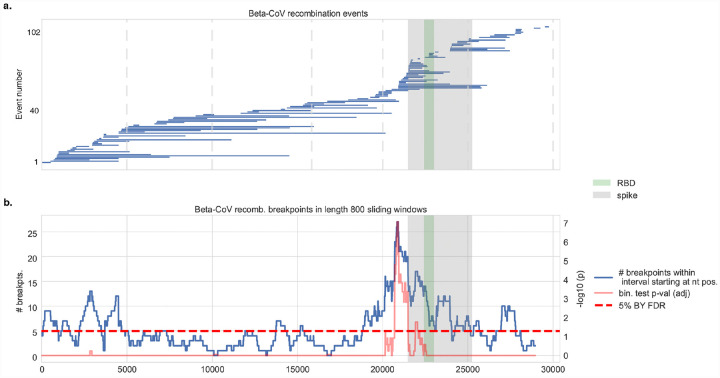
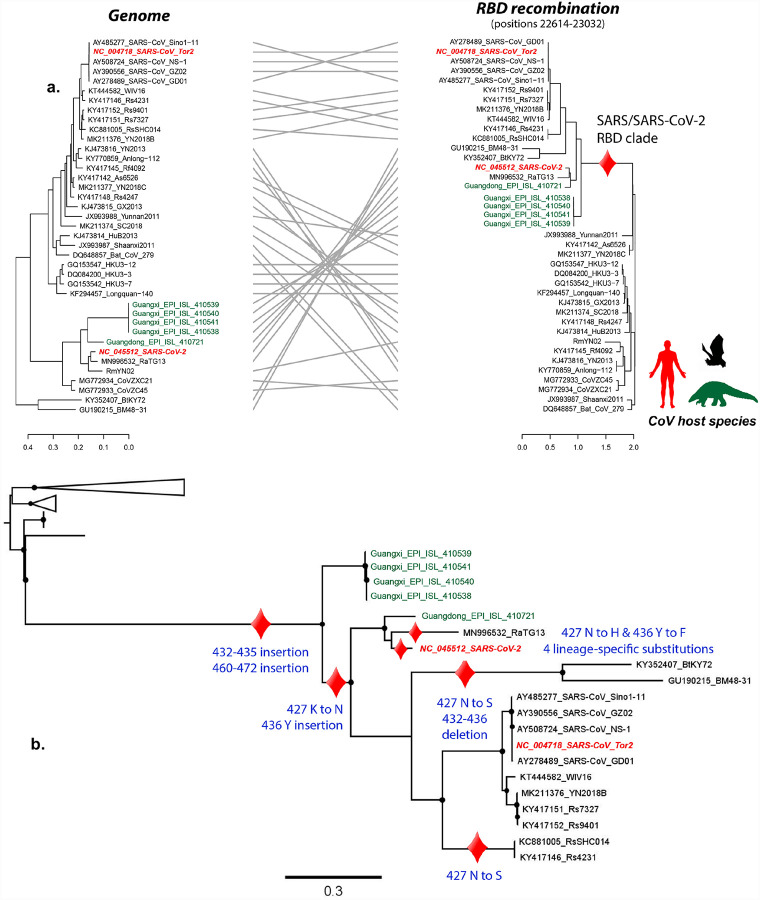
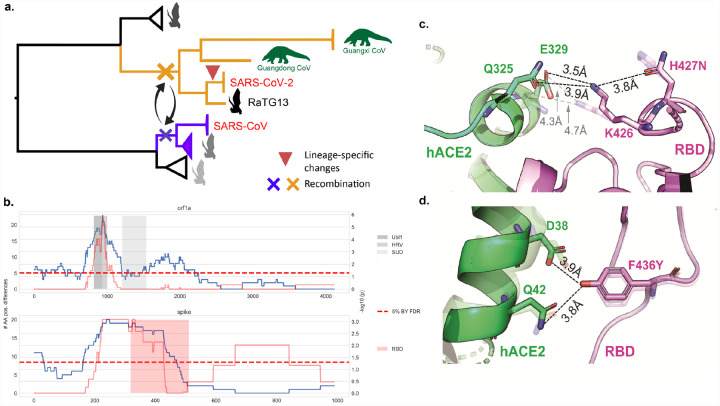
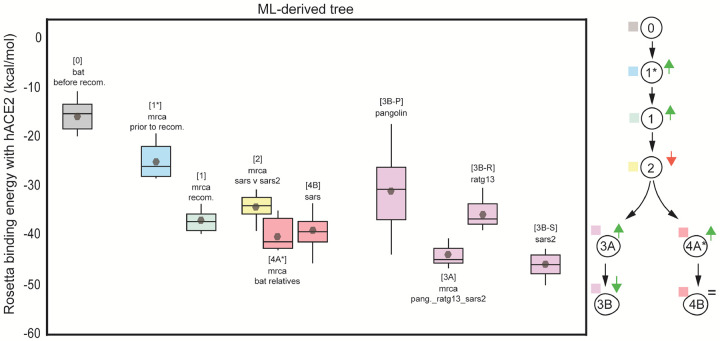
The emergence of SARS-CoV-2 underscores the need to better understand the evolutionary processes that drive the emergence and adaptation of zoonotic viruses in humans. In the betacoronavirus genus, which also includes SARS-CoV and MERS-CoV, recombination frequently encompasses the Receptor Binding Domain (RBD) of the Spike protein, which, in turn, is responsible for viral binding to host cell receptors. Here, we find evidence of a recombination event in the RBD involving ancestral linages to both SARS-CoV and SARS-CoV-2. Although we cannot specify the recombinant nor the parental strains, likely due to the ancestry of the event and potential undersampling, our statistical analyses in the space of phylogenetic trees support such an ancestral recombination. Consequently, SARS-CoV and SARS-CoV-2 share an RBD sequence that includes two insertions (positions 432-436 and 460-472), as well as the variants 427N and 436Y. Both 427N and 436Y belong to a helix that interacts directly with the human ACE2 (hACE2) receptor. Reconstruction of ancestral states, combined with protein-binding affinity analyses using the physics-based trRosetta algorithm, reveal that the recombination event involving ancestral strains of SARS-CoV and SARS-CoV-2 led to an increased affinity for hACE2 binding, and that alleles 427N and 436Y significantly enhanced affinity as well. Structural modeling indicates that ancestors of SARS-CoV-2 may have acquired the ability to infect humans decades ago. The binding affinity with the human receptor was subsequently boosted in SARS-CoV and SARS-CoV-2 through further mutations in RBD. In sum, we report an ancestral recombination event affecting the RBD of both SARS-CoV and SARS-CoV-2 that was associated with an increased binding affinity to hACE2.
Conflict of interest statement
Disclosure of Potential Conflicts of Interest R.R. is a member of the SAB of AimedBio in a project unrelated to the current manuscript. PKS is a member of the SAB or Board of Directors of Applied Biomath LLC, Glencoe Software Inc, and RareCyte Inc and has equity in these companies; his is also on the SAB of NanoString Inc. In the last five years the Sorger lab has received research funding from Novartis and Merck. Sorger declares that none of these relationships are directly or indirectly related to the content of this manuscript. The other authors declare no conflicts.
Figures


References
-
- W.H.O. Coronavirus disease 2019 (COVID-19) Situation Report - 5 January. (2021).
-
- Peng Zhou, X.-L. Y., Wang Xian-Guang, Hu Ben, Zhang Lei, Zhang Wei, Si Hao-Rui, Zhu Yan, Li Bei, Huang Chao-Lin, Chen Hui-Dong, Chen Jing, Luo Yun, Guo Hua, Jiang Ren-Di, Liu Mei-Qin, Chen Ying, Shen Xu-Rui, Wang Xi, Zheng Xiao-Shuang, Zhao Kai, Chen Quan-Jiao, Deng Fei, Liu Lin-Lin, Yan Bing, Zhan Fa-Xian, Wang Yan-Yi, Xiao Gengfu, Shi Zheng-Li. Discovery of a novel coronavirus associated with the recent pneumonia outbreak in humans and its potential bat origin. bioRxiv 2020.01.22.914952 doi: 10.1101/2020.01.22.914952 (2020). - DOI
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous