

This is a preprint.
The emergence of SARS-CoV-2 in Europe and the US
- PMID: 32511416
- PMCID: PMC7265688
- DOI: 10.1101/2020.05.21.109322
The emergence of SARS-CoV-2 in Europe and the US
Update in
-
The emergence of SARS-CoV-2 in Europe and North America.Science. 2020 Oct 30;370(6516):564-570. doi: 10.1126/science.abc8169. Epub 2020 Sep 10. Science. 2020. PMID: 32912998 Free PMC article.
Abstract
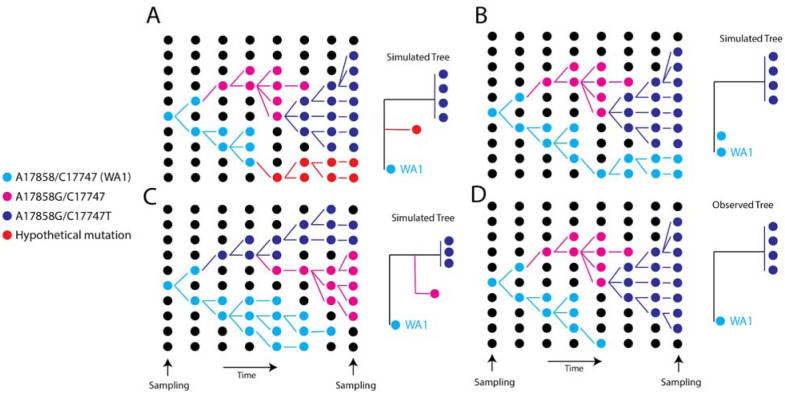
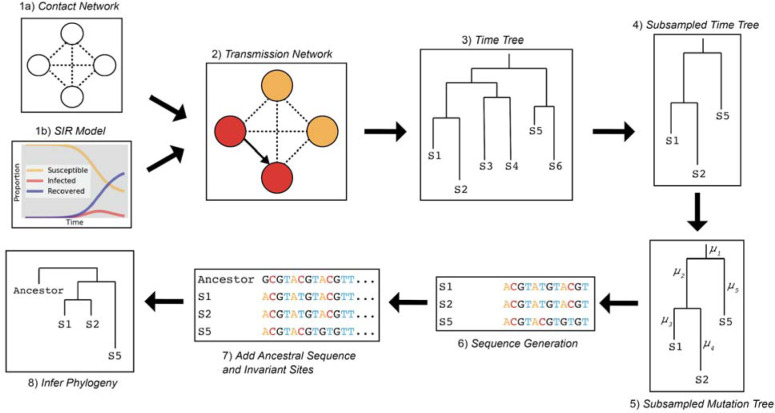
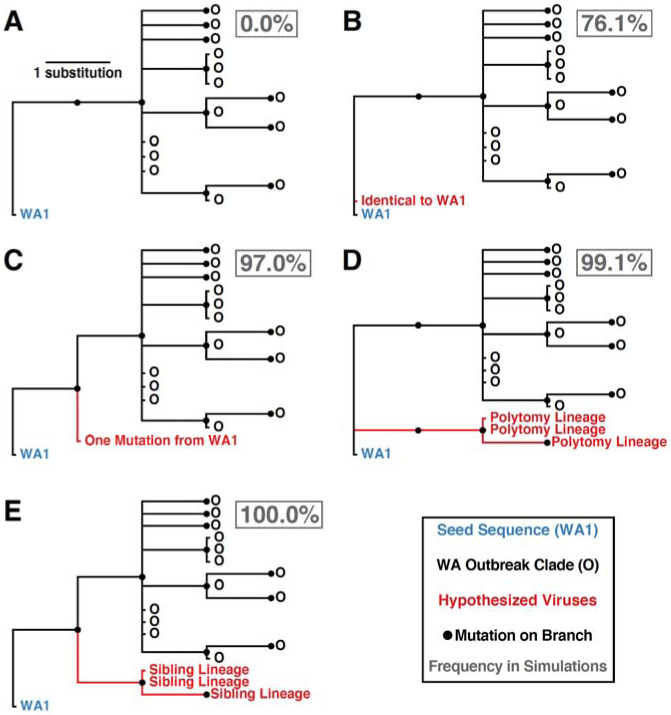
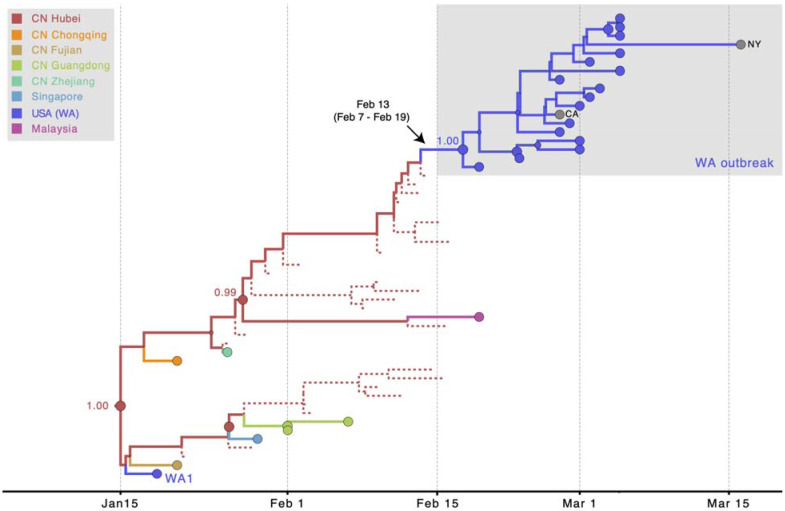
Accurate understanding of the global spread of emerging viruses is critically important for public health response and for anticipating and preventing future outbreaks. Here, we elucidate when, where and how the earliest sustained SARS-CoV-2 transmission networks became established in Europe and the United States (US). Our results refute prior findings erroneously linking cases in January 2020 with outbreaks that occurred weeks later. Instead, rapid interventions successfully prevented onward transmission of those early cases in Germany and Washington State. Other, later introductions of the virus from China to both Italy and Washington State founded the earliest sustained European and US transmission networks. Our analyses reveal an extended period of missed opportunity when intensive testing and contact tracing could have prevented SARS-CoV-2 from becoming established in the US and Europe.
Conflict of interest statement
Competing Interests: JOW has received funding from Gilead Sciences, LLC (completed) and the CDC (ongoing) via grants and contracts to his institution unrelated to this research. MAS receives funding from Janssen Research & Development, IQVIA and Private Health Management via contracts unrelated to this research.
Figures

References
-
- Wu F., Zhao S., Yu B., Chen Y.-M., Wang W., Song Z.-G., Hu Y., Tao Z.-W., Tian J.-H., Pei Y.-Y., Yuan M.-L., Zhang Y.-L., Dai F.-H., Liu Y., Wang Q.-M., Zheng J.-J., Xu L., Holmes E. C., Zhang Y.-Z., A new coronavirus associated with human respiratory disease in China. Nature. 579, 265–269 (2020). - PMC - PubMed
-
- Holshue M. L., DeBolt C., Lindquist S., Lofy K. H., Wiesman J., Bruce H., Spitters C., Ericson K., Wilkerson S., Tural A., Diaz G., Cohn A., Fox L., Patel A., Gerber S. I., Kim L., Tong S., Lu X., Lindstrom S., Pallansch M. A., Weldon W. C., Biggs H. M., Uyeki T. M., Pillai S. K., Washington State 2019-nCoV Case Investigation Team, First Case of 2019 Novel Coronavirus in the United States. N. Engl. J. Med. 382, 929–936 (2020). - PMC - PubMed
-
- Harmon A., Inside the Race to Contain America’s First Coronavirus Case. The New York Times (2020), (available at https://www.nytimes.com/2020/02/05/us/corona-virus-washington-state.html).
-
- T. B. (@trvrb), The team at the @seattleflustudy have sequenced the genome the #COVID19 community case reported yesterday from Snohomish County, WA, and have posted the sequence publicly to http://gisaid.org. There are some enormous implications here. Twitter (2020), (available at https://twitter.com/trvrb/status/1233970271318503426).
-
- Chu H. Y., Englund J. A., Starita L. M., Famulare M., Brandstetter E., Nickerson D. A., Rieder M. J., Adler A., Lacombe K., Kim A. E., Graham C., Logue J., Wolf C. R., Heimonen J., McCulloch D. J., Han P. D., Sibley T. R., Lee J., Ilcisin M., Fay K., Burstein R., Martin B., Lockwood C. M., Thompson M., Lutz B., Jackson M., Hughes J. P., Boeckh M., Shendure J., Bedford T., Seattle Flu Study Investigators, Early Detection of Covid-19 through a Citywide Pandemic Surveillance Platform. N. Engl. J. Med. (2020), doi:10.1056/NEJMc2008646 - DOI - PMC - PubMed
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous