

Divergence, Convergence, and Therapeutic Implications: A Cell Biology Perspective of C9ORF72-ALS/FTD
- PMID: 32513219
- PMCID: PMC7282082
- DOI: 10.1186/s13024-020-00383-7
Divergence, Convergence, and Therapeutic Implications: A Cell Biology Perspective of C9ORF72-ALS/FTD
Erratum in
-
Correction to: Divergence, Convergence, and Therapeutic Implications: A Cell Biology Perspective of C9ORF72-ALS/FTD.Mol Neurodegener. 2020 Jul 1;15(1):37. doi: 10.1186/s13024-020-00390-8. Mol Neurodegener. 2020. PMID: 32611370 Free PMC article.
Abstract
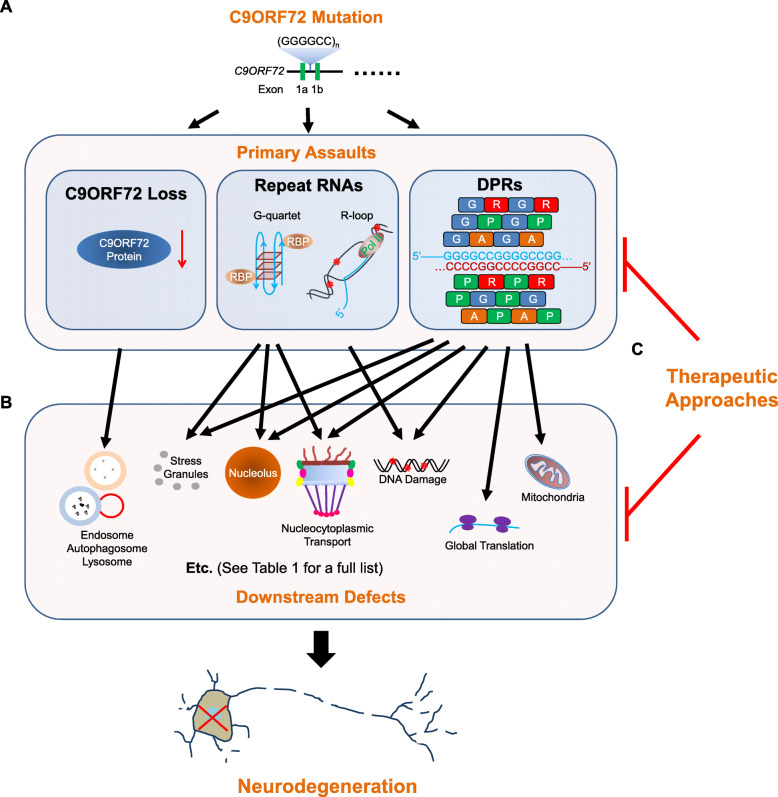
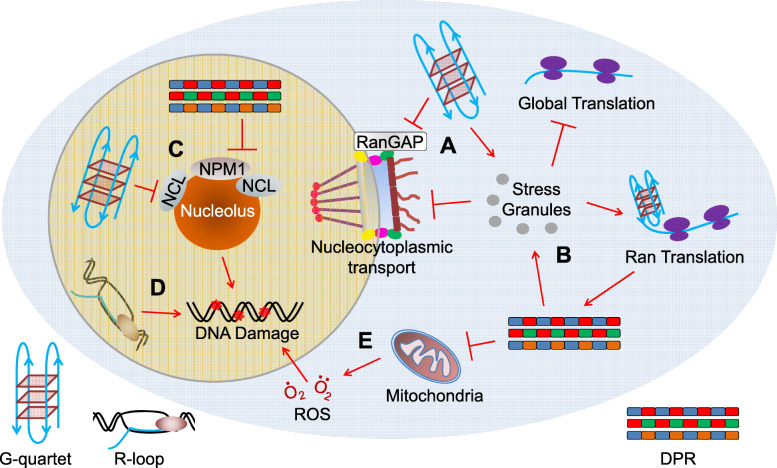
Ever since a GGGGCC hexanucleotide repeat expansion mutation in C9ORF72 was identified as the most common cause of familial amyotrophic lateral sclerosis (ALS) and frontotemporal dementia (FTD), three competing but nonexclusive hypotheses to explain how this mutation causes diseases have been proposed and are still under debate. Recent studies in the field have tried to understand how the repeat expansion disrupts cellular physiology, which has suggested interesting convergence of these hypotheses on downstream, functional defects in cells, such as nucleocytoplasmic transport disruption, membrane-less organelle defects, and DNA damage. These studies have not only provided an integrated view of the disease mechanism but also revealed novel cell biology implicated in neurodegeneration. Furthermore, some of the discoveries have given rise to new ideas for therapeutic development. Here, we review the research progress on cellular pathophysiology of C9ORF72-mediated ALS and FTD and its therapeutic implication. We suggest that the repeat expansion drives pathogenesis through a combination of downstream defects, of which some can be therapeutic targets.
Keywords: Amyotrophic lateral sclerosis; C9orf72; Frontotemporal dementia.
Conflict of interest statement
B.O. is on the advisory board of MediciNova and Biogen Idec. The other authors declare no competing interests.
Figures
References
-
- Neary D, Snowden JS, Gustafson L, Passant U, Stuss D, Black S, Freedman M, Kertesz A, Robert PH, Albert M, et al. Frontotemporal lobar degeneration: a consensus on clinical diagnostic criteria. Neurology. 1998;51:1546–1554. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
