

Topoisomerase I-driven repair of UV-induced damage in NER-deficient cells
- PMID: 32513688
- PMCID: PMC7321995
- DOI: 10.1073/pnas.1920165117
Topoisomerase I-driven repair of UV-induced damage in NER-deficient cells
Abstract
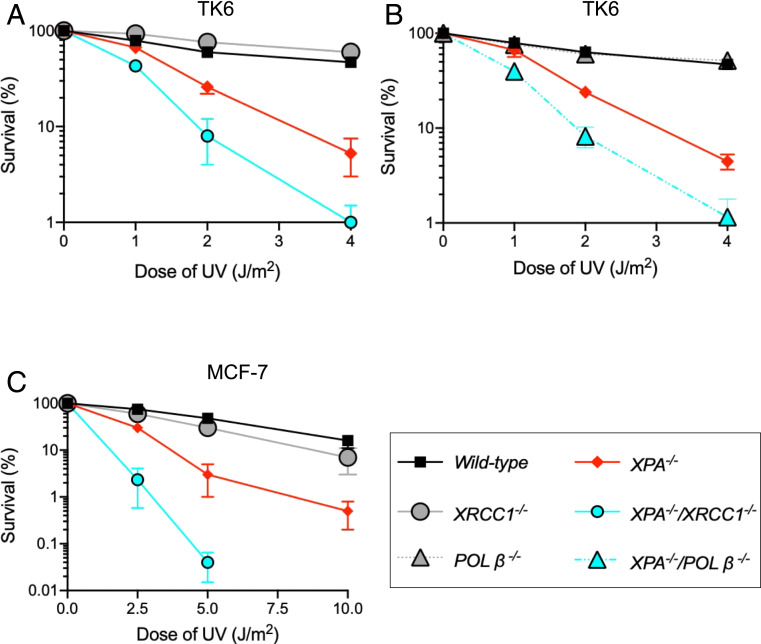
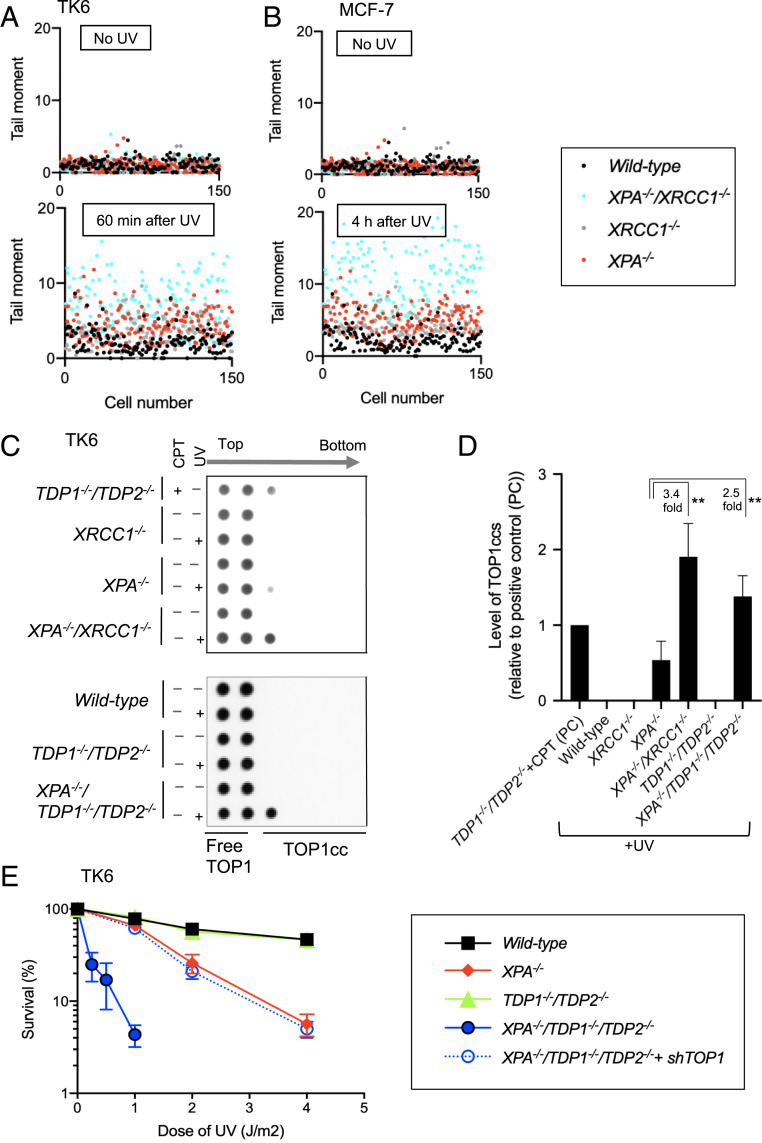
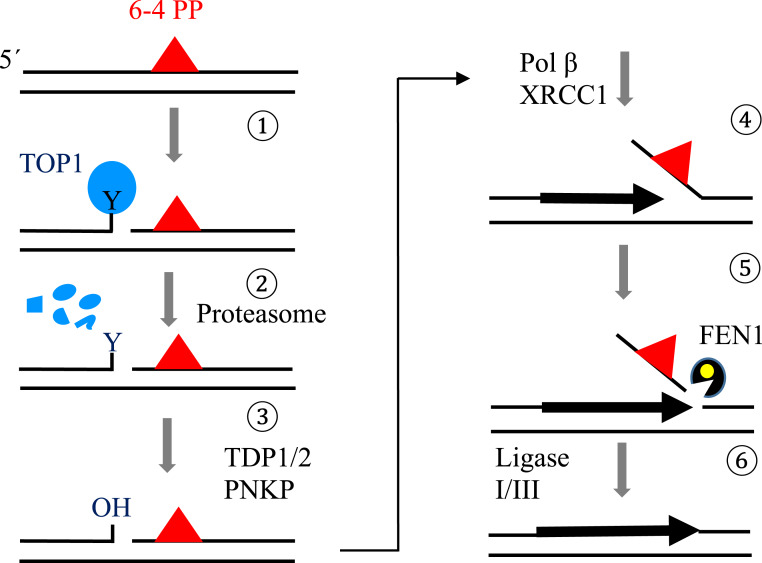
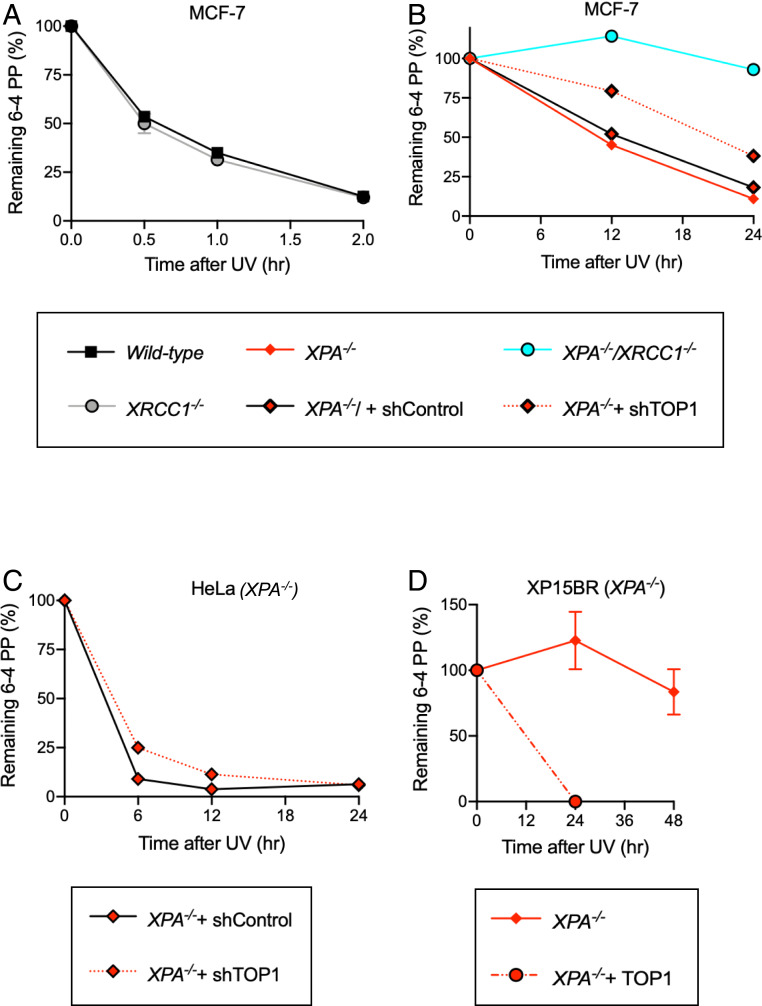
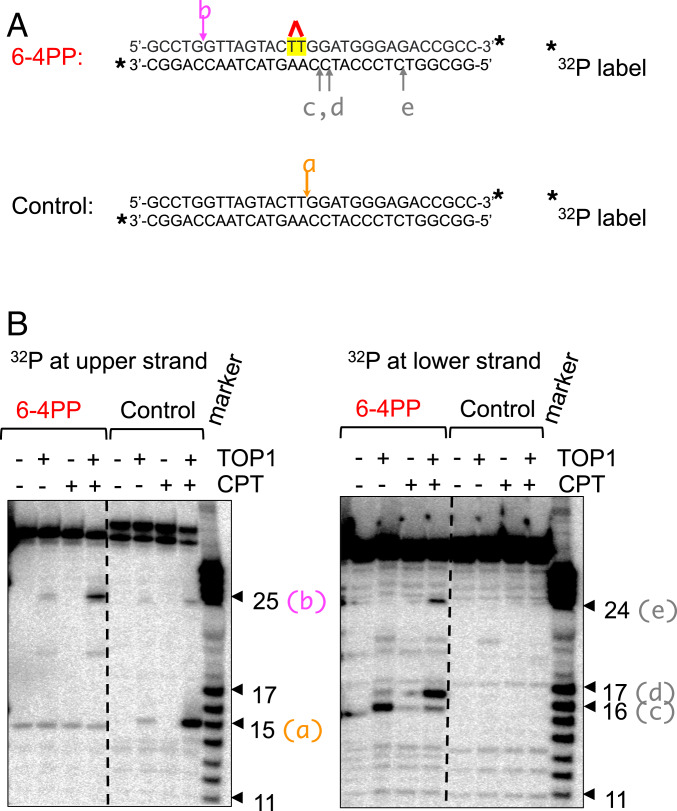
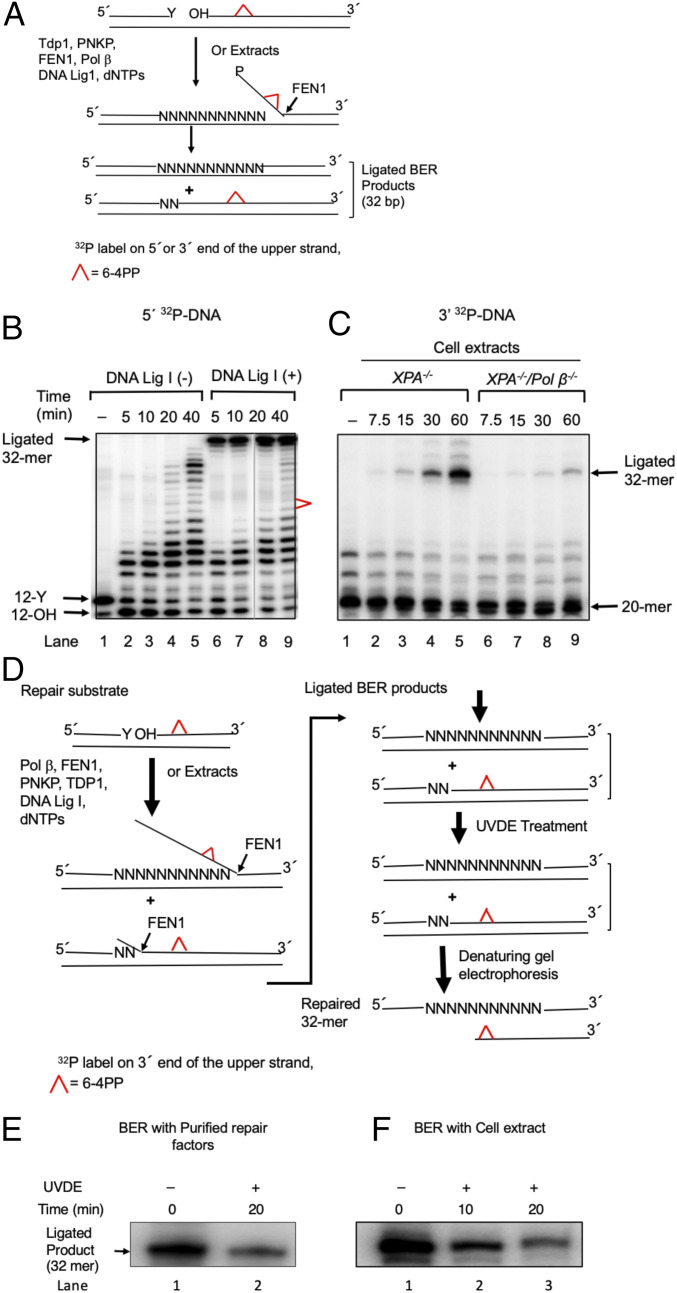
Nucleotide excision repair (NER) removes helix-destabilizing adducts including ultraviolet (UV) lesions, cyclobutane pyrimidine dimers (CPDs), and pyrimidine (6-4) pyrimidone photoproducts (6-4PPs). In comparison with CPDs, 6-4PPs have greater cytotoxicity and more strongly destabilizing properties of the DNA helix. It is generally believed that NER is the only DNA repair pathway that removes the UV lesions as evidenced by the previous data since no repair of UV lesions was detected in NER-deficient skin fibroblasts. Topoisomerase I (TOP1) constantly creates transient single-strand breaks (SSBs) releasing the torsional stress in genomic duplex DNA. Stalled TOP1-SSB complexes can form near DNA lesions including abasic sites and ribonucleotides embedded in chromosomal DNA. Here we show that base excision repair (BER) increases cellular tolerance to UV independently of NER in cancer cells. UV lesions irreversibly trap stable TOP1-SSB complexes near the UV damage in NER-deficient cells, and the resulting SSBs activate BER. Biochemical experiments show that 6-4PPs efficiently induce stable TOP1-SSB complexes, and the long-patch repair synthesis of BER removes 6-4PPs downstream of the SSB. Furthermore, NER-deficient cancer cell lines remove 6-4PPs within 24 h, but not CPDs, and the removal correlates with TOP1 expression. NER-deficient skin fibroblasts weakly express TOP1 and show no detectable repair of 6-4PPs. Remarkably, the ectopic expression of TOP1 in these fibroblasts led them to completely repair 6-4PPs within 24 h. In conclusion, we reveal a DNA repair pathway initiated by TOP1, which significantly contributes to cellular tolerance to UV-induced lesions particularly in malignant cancer cells overexpressing TOP1.
Keywords: 6–4PPs; UV damage; base excision repair; topoisomerase I.
Conflict of interest statement
The authors declare no competing interest.
Figures
References
-
- Mitchell D. L., The induction and repair of lesions produced by the photolysis of (6-4) photoproducts in normal and UV-hypersensitive human cells. Mutat. Res. 194, 227–237 (1988). - PubMed
-
- Mitchell D. L., The relative cytotoxicity of (6-4) photoproducts and cyclobutane dimers in mammalian cells. Photochem. Photobiol. 48, 51–57 (1988). - PubMed
-
- Costa R. M., Chiganças V., Galhardo R. d S., Carvalho H., Menck C. F., The eukaryotic nucleotide excision repair pathway. Biochimie 85, 1083–1099 (2003). - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
