

Large-scale genome-wide association study in a Japanese population identifies novel susceptibility loci across different diseases
- PMID: 32514122
- PMCID: PMC7968075
- DOI: 10.1038/s41588-020-0640-3
Large-scale genome-wide association study in a Japanese population identifies novel susceptibility loci across different diseases
Abstract
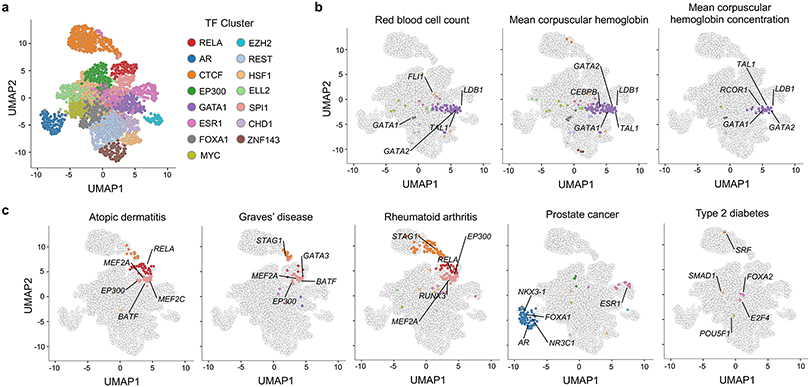
The overwhelming majority of participants in current genetic studies are of European ancestry. To elucidate disease biology in the East Asian population, we conducted a genome-wide association study (GWAS) with 212,453 Japanese individuals across 42 diseases. We detected 320 independent signals in 276 loci for 27 diseases, with 25 novel loci (P < 9.58 × 10-9). East Asian-specific missense variants were identified as candidate causal variants for three novel loci, and we successfully replicated two of them by analyzing independent Japanese cohorts; p.R220W of ATG16L2 (associated with coronary artery disease) and p.V326A of POT1 (associated with lung cancer). We further investigated enrichment of heritability within 2,868 annotations of genome-wide transcription factor occupancy, and identified 378 significant enrichments across nine diseases (false discovery rate < 0.05) (for example, NKX3-1 for prostate cancer). This large-scale GWAS in a Japanese population provides insights into the etiology of complex diseases and highlights the importance of performing GWAS in non-European populations.
Figures













References
-
- Diversity matters. Nature Reviews Genetics 20, 495 (2019). - PubMed
REFERENCES (for method)
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
