

TETs compete with DNMT3 activity in pluripotent cells at thousands of methylated somatic enhancers
- PMID: 32514123
- PMCID: PMC7415576
- DOI: 10.1038/s41588-020-0639-9
TETs compete with DNMT3 activity in pluripotent cells at thousands of methylated somatic enhancers
Abstract
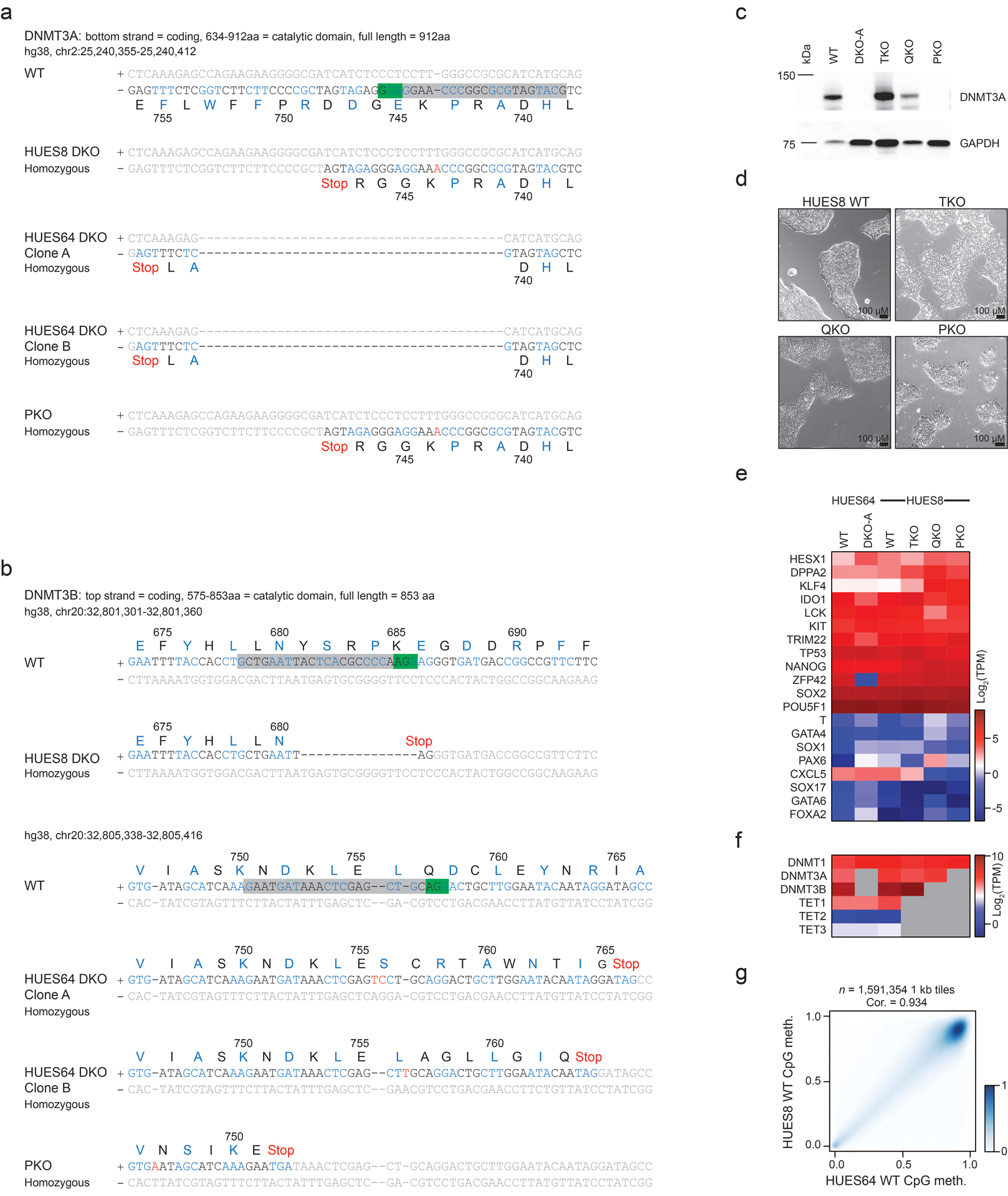
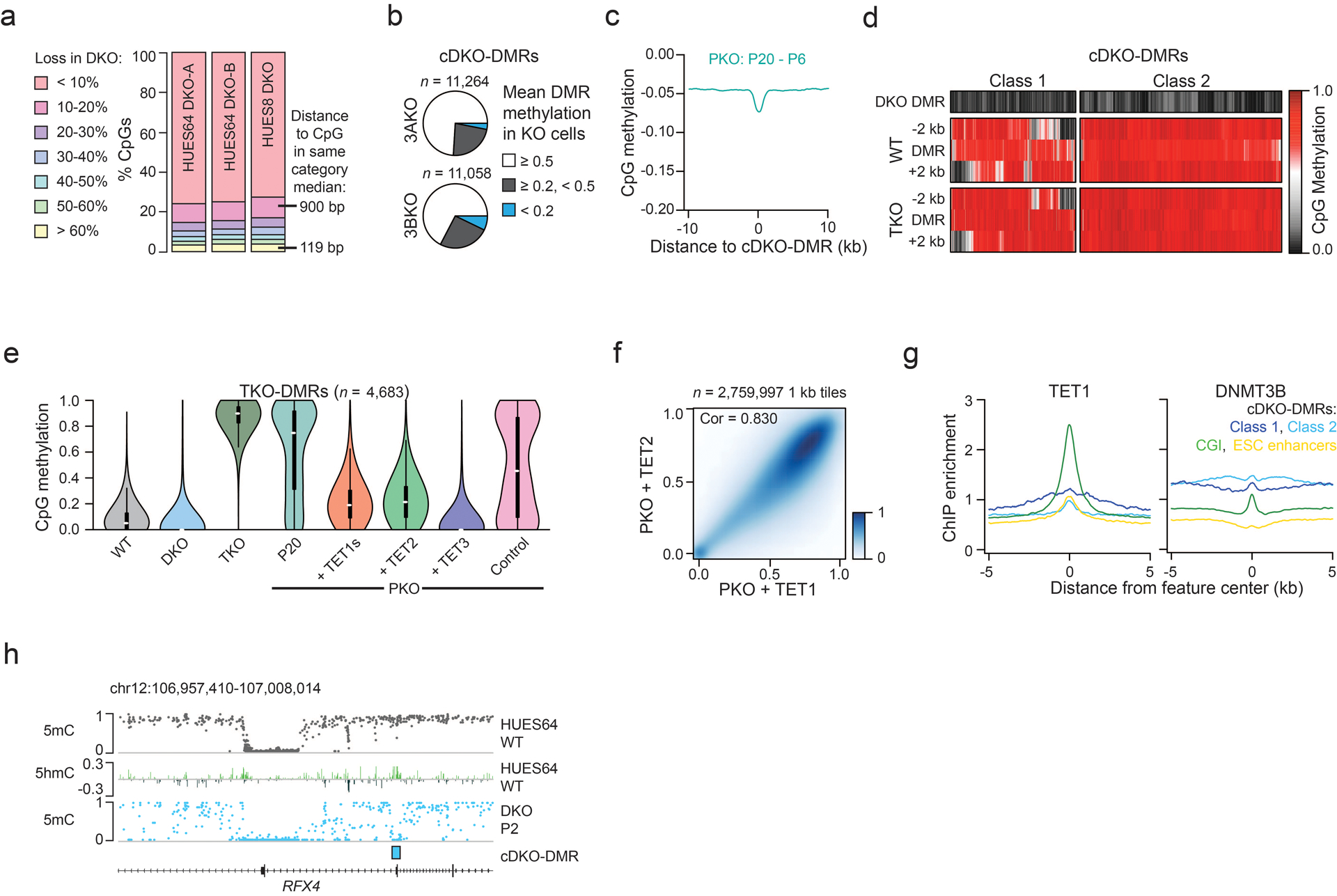
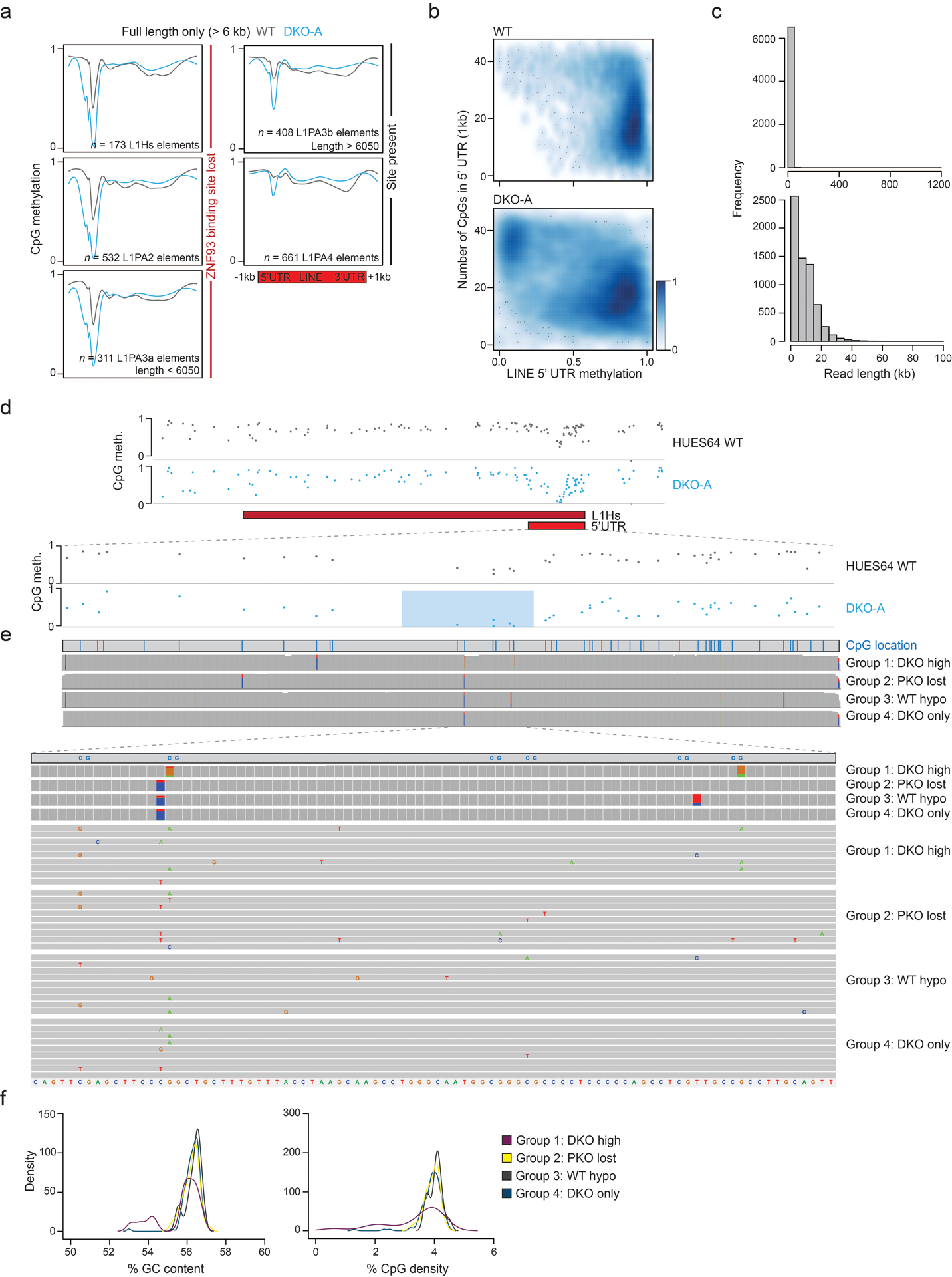
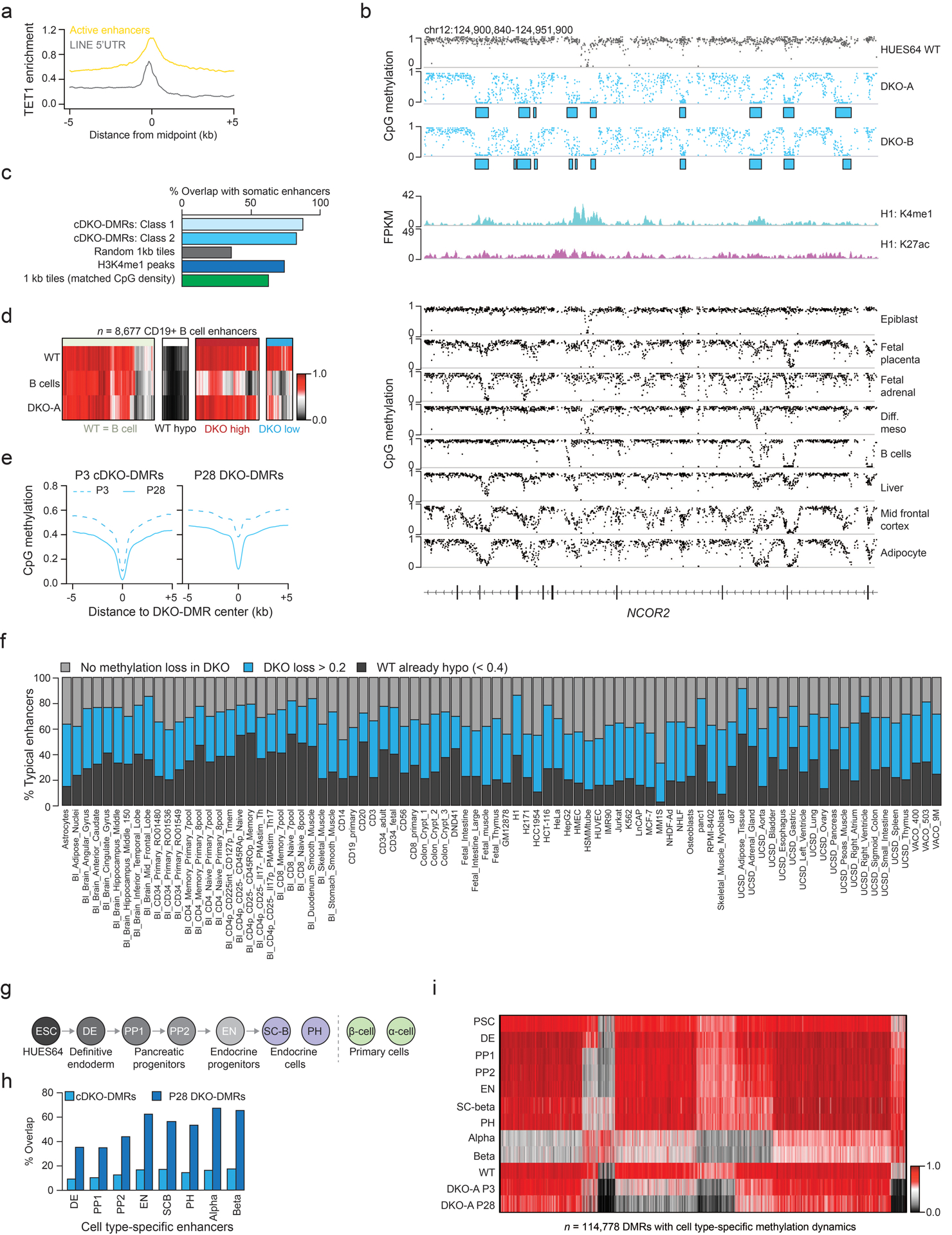
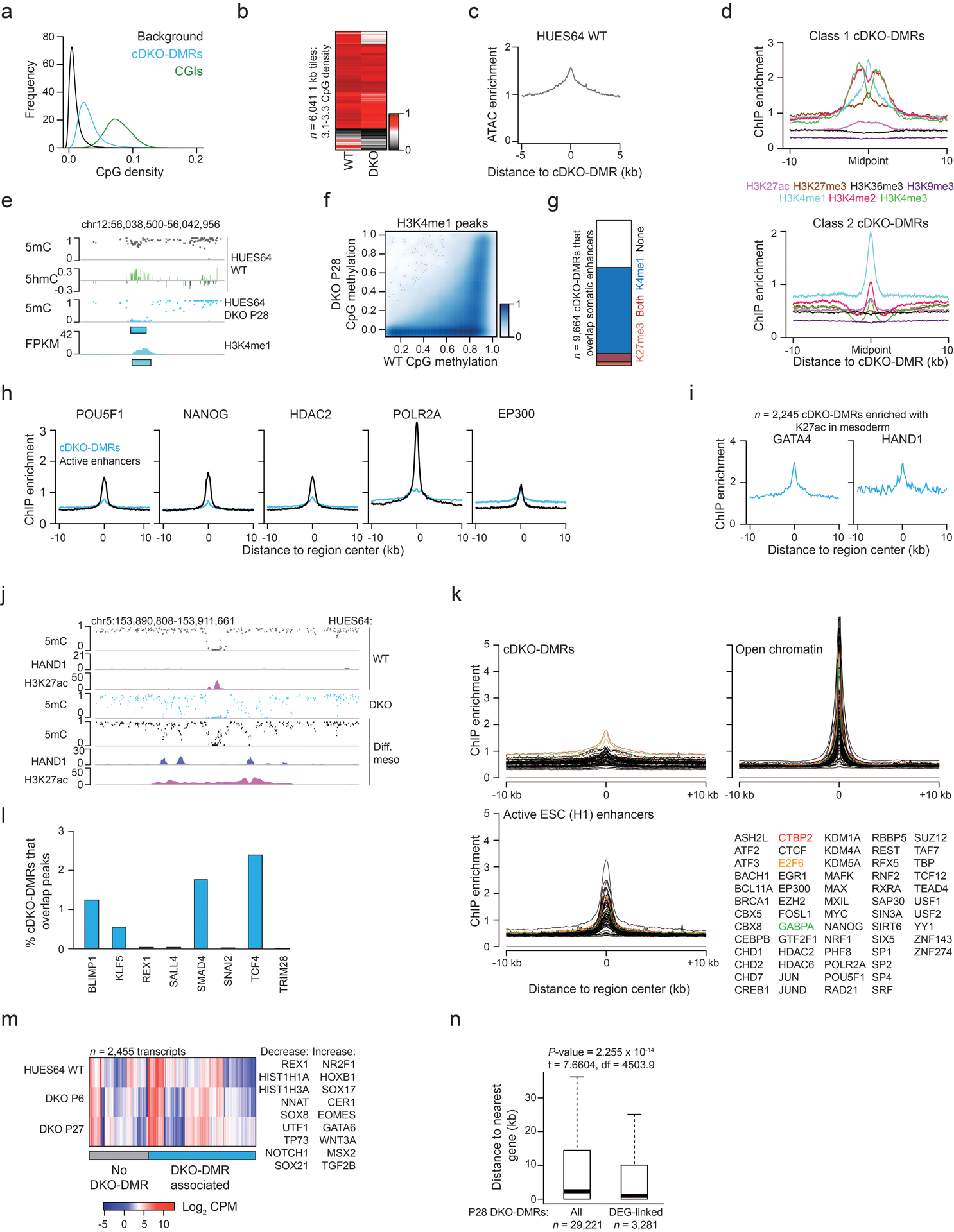
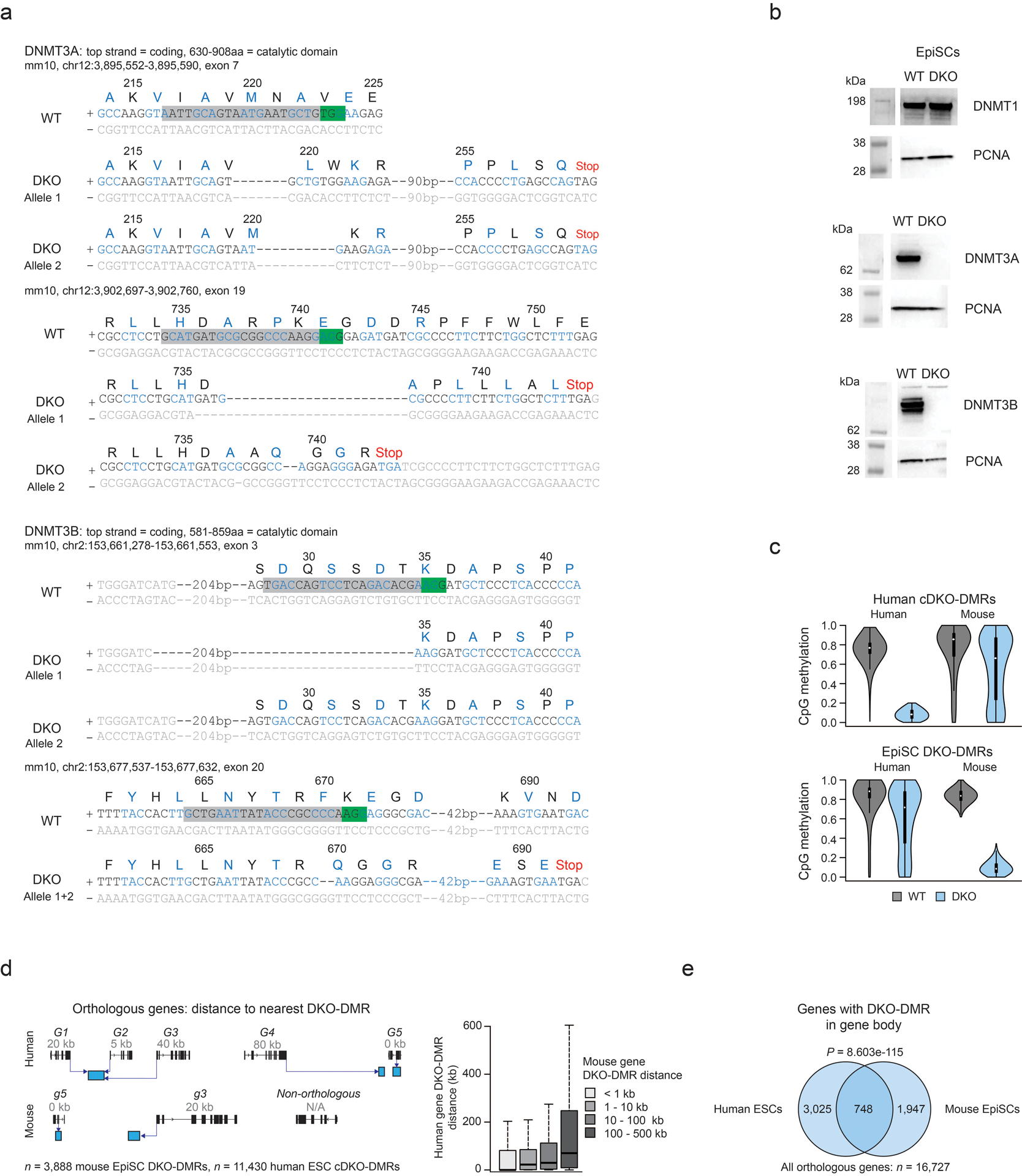
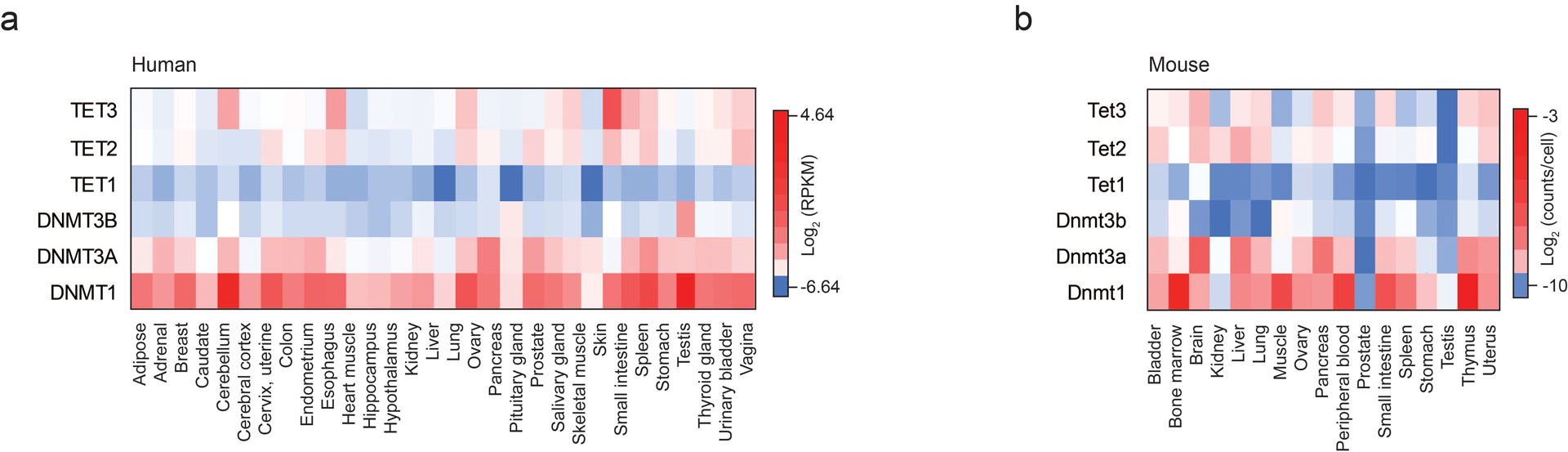
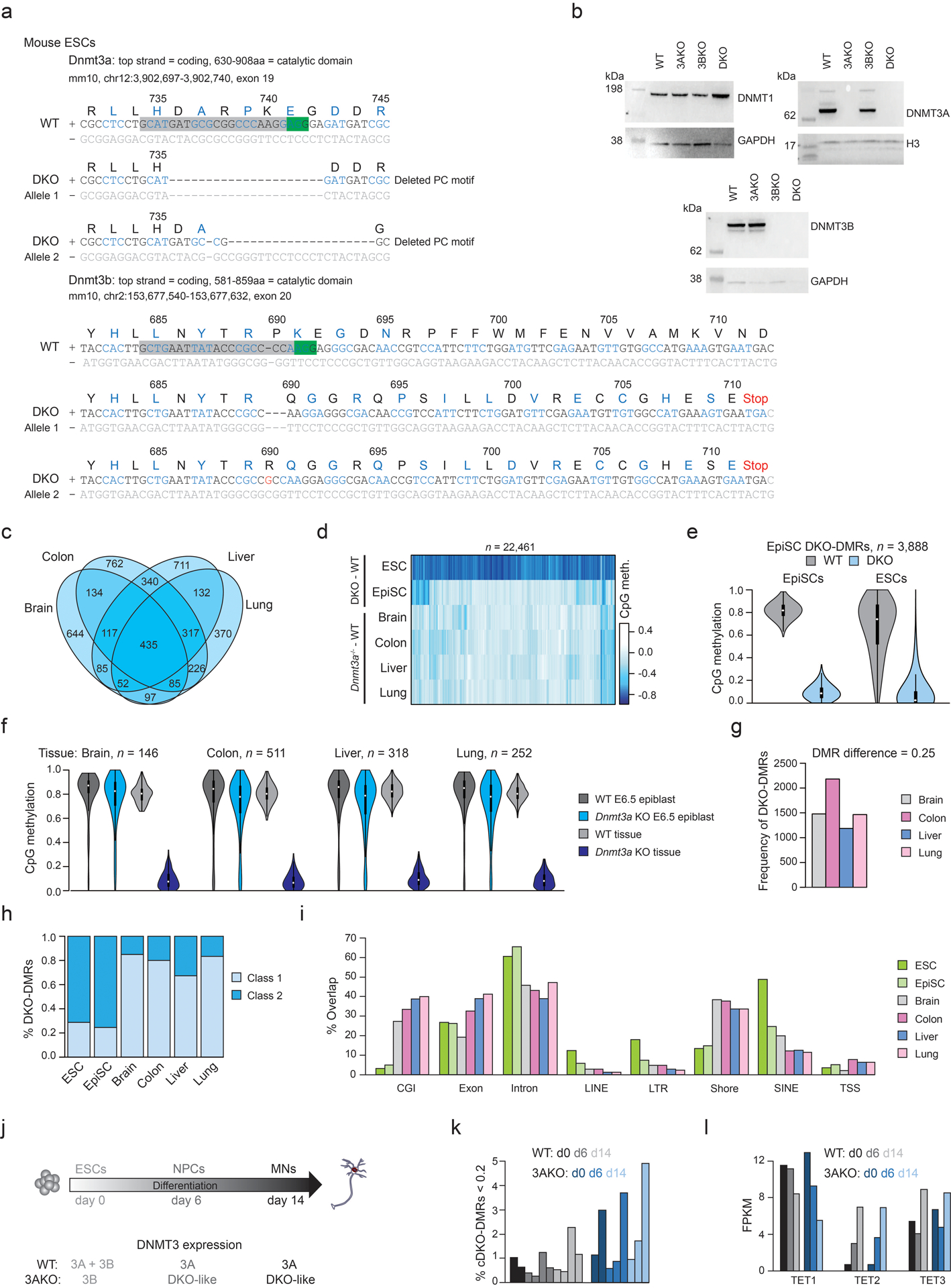
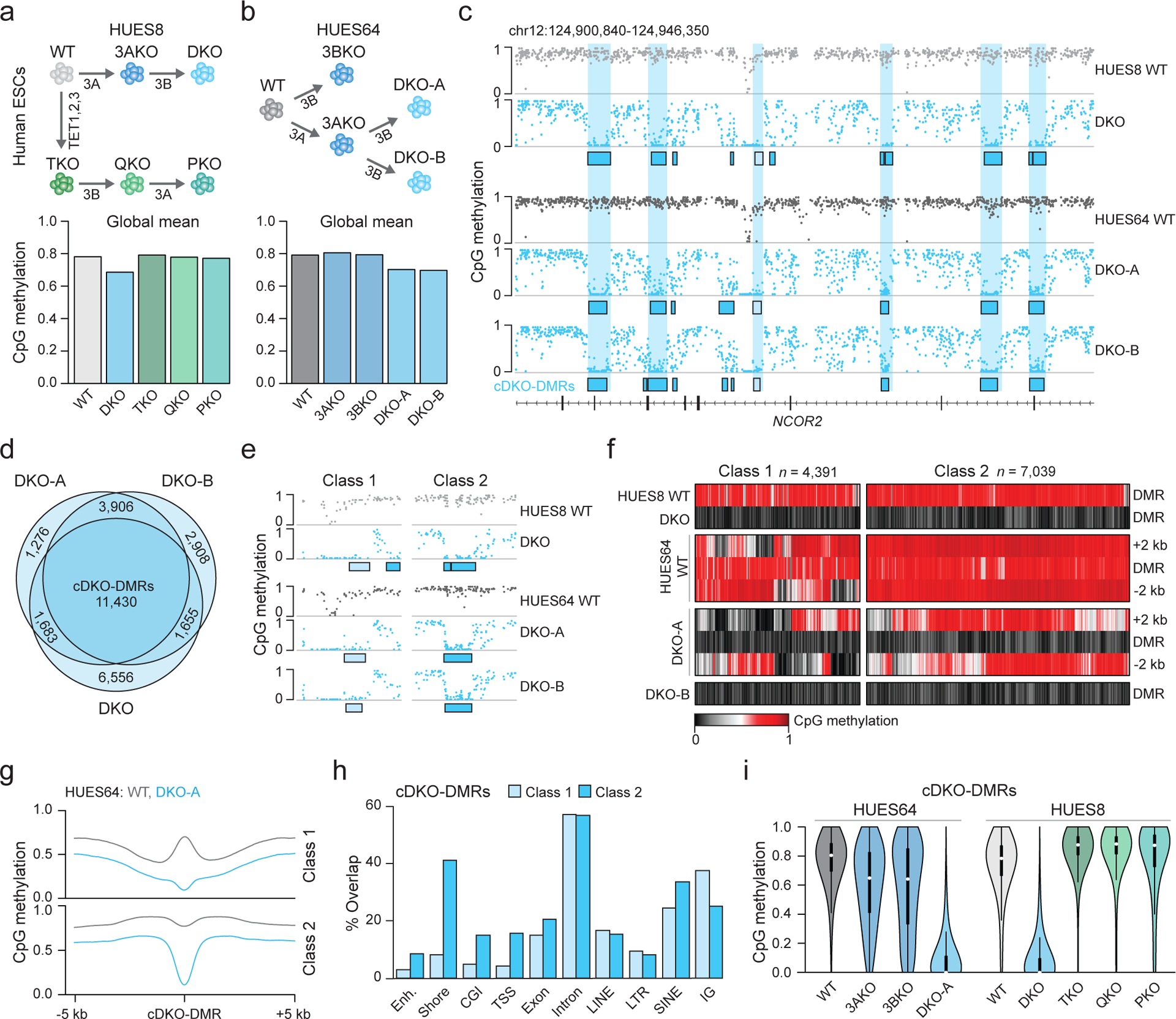
Mammalian cells stably maintain high levels of DNA methylation despite expressing both positive (DNMT3A/B) and negative (TET1-3) regulators. Here, we analyzed the independent and combined effects of these regulators on the DNA methylation landscape using a panel of knockout human embryonic stem cell (ESC) lines. The greatest impact on global methylation levels was observed in DNMT3-deficient cells, including reproducible focal demethylation at thousands of normally methylated loci. Demethylation depends on TET expression and occurs only when both DNMT3s are absent. Dynamic loci are enriched for hydroxymethylcytosine and overlap with subsets of putative somatic enhancers that are methylated in ESCs and can be activated upon differentiation. We observe similar dynamics in mouse ESCs that were less frequent in epiblast stem cells (EpiSCs) and scarce in somatic tissues, suggesting a conserved pluripotency-linked mechanism. Taken together, our data reveal tightly regulated competition between DNMT3s and TETs at thousands of somatic regulatory sequences within pluripotent cells.
Conflict of interest statement
Competing interests statement
The authors declare no competing interests.
Figures






References
-
- Gangisetty O, Cabrera MA & Murugan S Impact of epigenetics in aging and age related neurodegenerative diseases. Frontiers in bioscience (Landmark edition) 23, 1445–1464 (2018). - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
