

MET-dependent solid tumours - molecular diagnosis and targeted therapy
- PMID: 32514147
- PMCID: PMC7478851
- DOI: 10.1038/s41571-020-0377-z
MET-dependent solid tumours - molecular diagnosis and targeted therapy
Abstract
Attempts to develop MET-targeted therapies have historically focused on MET-expressing cancers, with limited success. Thus, MET expression in the absence of a genomic marker of MET dependence is a poor predictor of benefit from MET-targeted therapy. However, owing to the development of more sensitive methods of detecting genomic alterations, high-level MET amplification and activating MET mutations or fusions are all now known to be drivers of oncogenesis. MET mutations include those affecting the kinase or extracellular domains and those that result in exon 14 skipping. The activity of MET tyrosine kinase inhibitors varies by MET alteration category. The likelihood of benefit from MET-targeted therapies increases with increasing levels of MET amplification, although no consensus exists on the optimal diagnostic cut-off point for MET copy number gains identified using fluorescence in situ hybridization and, in particular, next-generation sequencing. Several agents targeting exon 14 skipping alterations are currently in clinical development, with promising data available from early-phase trials. By contrast, the therapeutic implications of MET fusions remain underexplored. Here we summarize and evaluate the utility of various diagnostic techniques and the roles of different classes of MET-targeted therapies in cancers with MET amplification, mutation and fusion, and MET overexpression.
Figures
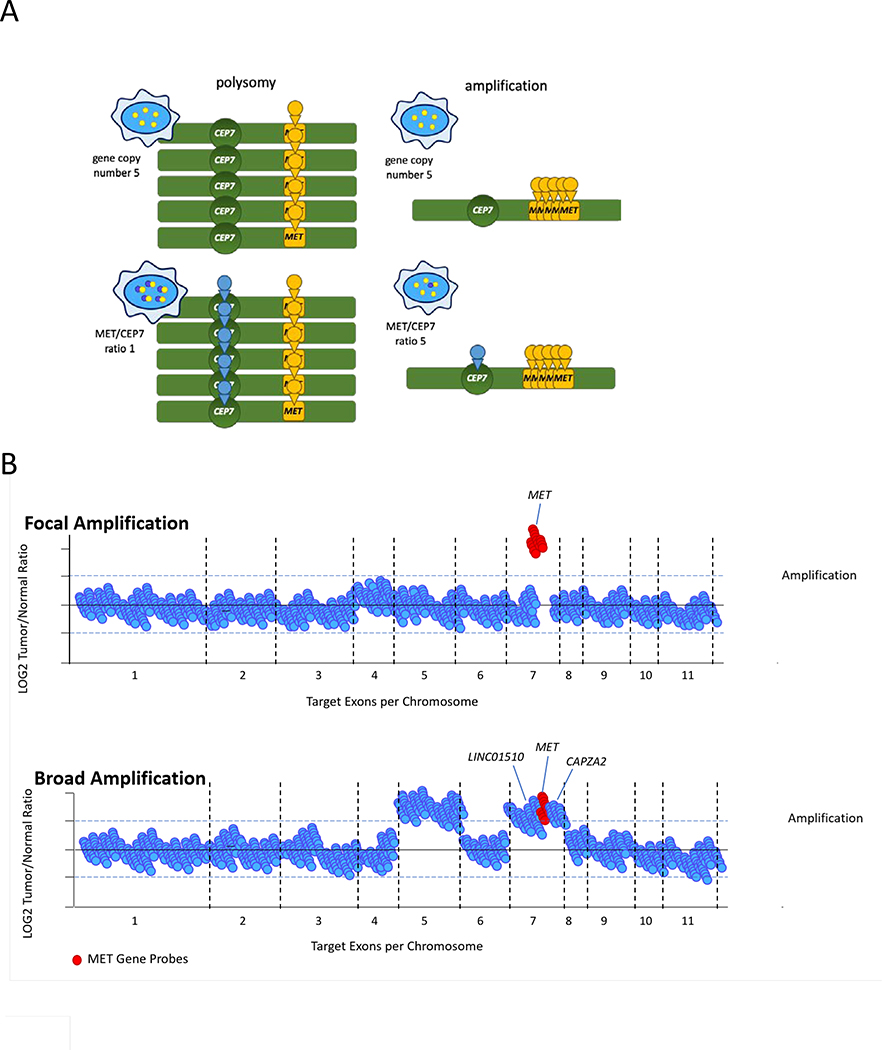

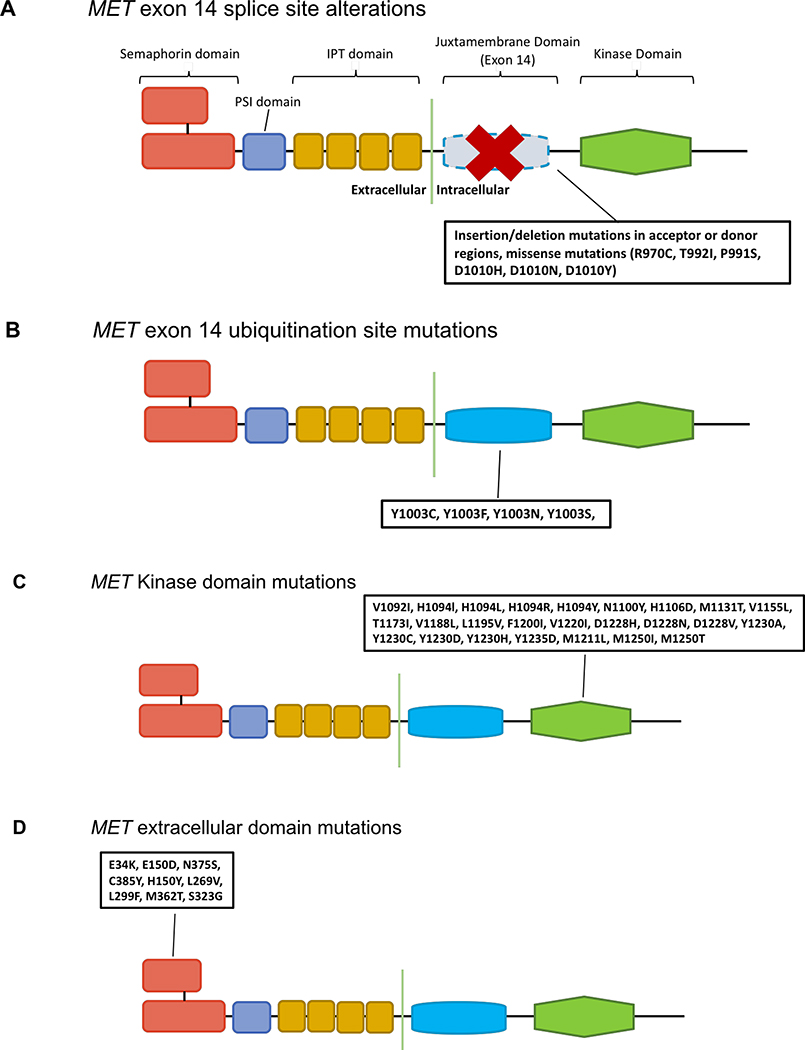
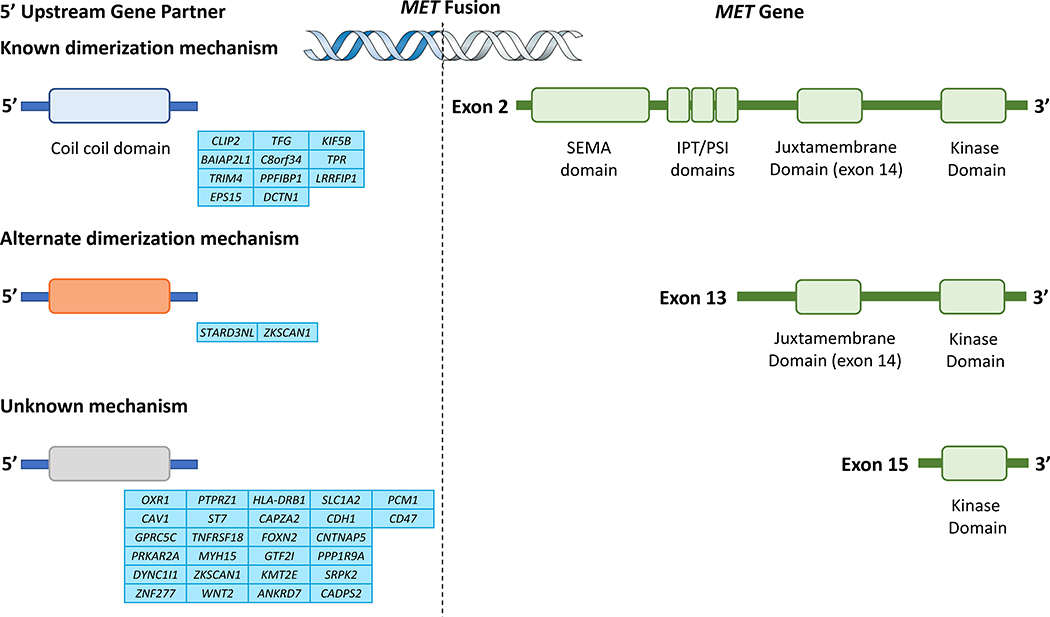
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
