

NormQ: RNASeq normalization based on RT-qPCR derived size factors
- PMID: 32514328
- PMCID: PMC7264052
- DOI: 10.1016/j.csbj.2020.05.010
NormQ: RNASeq normalization based on RT-qPCR derived size factors
Abstract
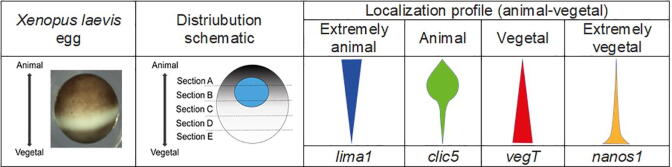
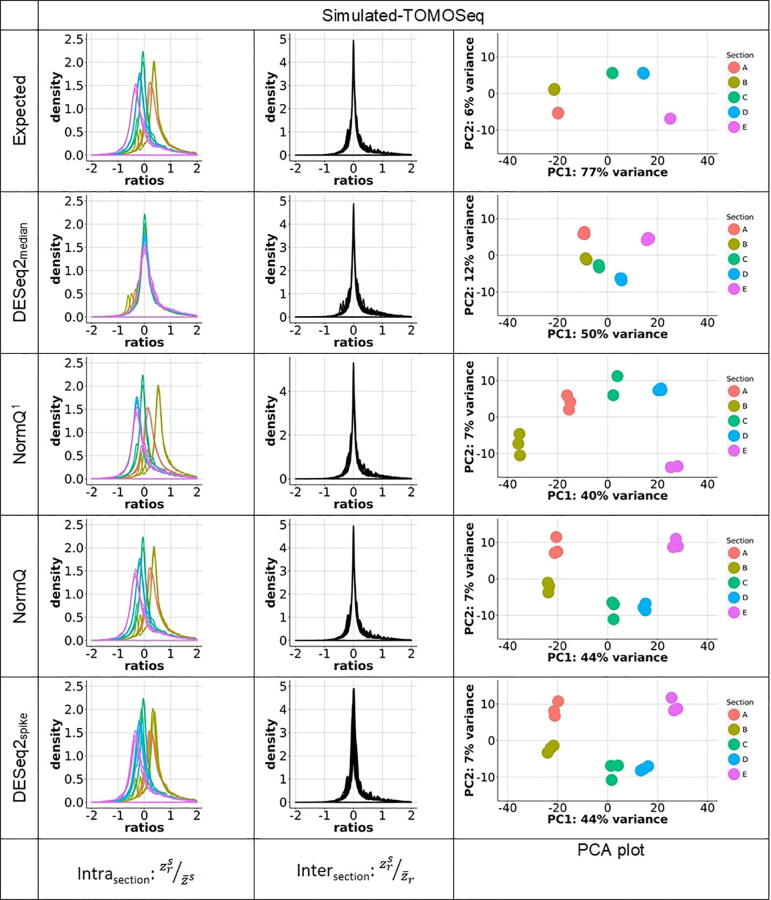
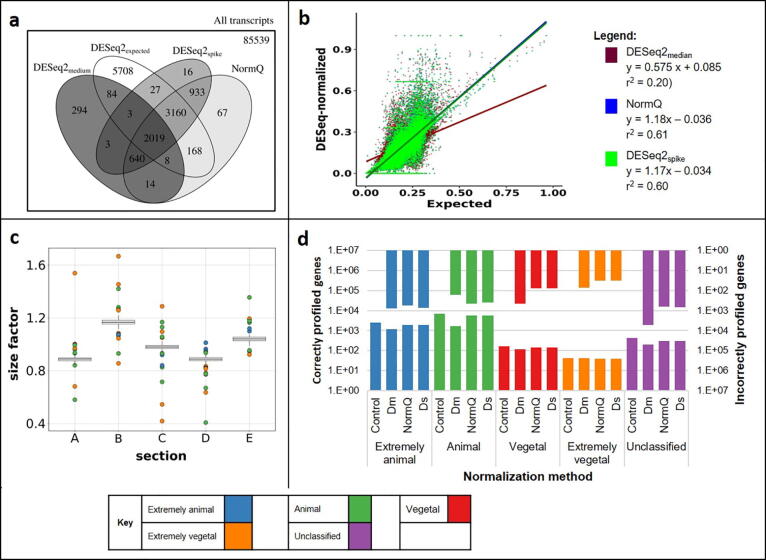
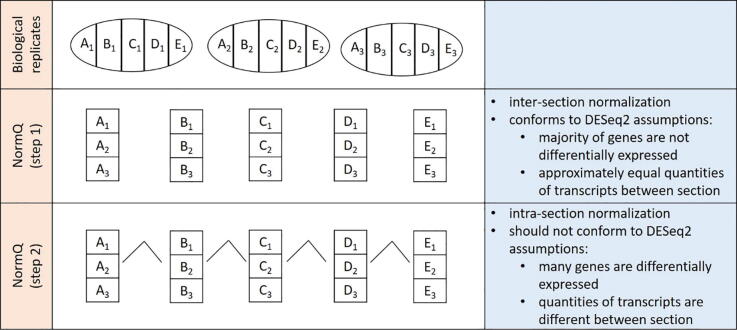
The merit of RNASeq data relies heavily on correct normalization. However, most methods assume that the majority of transcripts show no differential expression between conditions. This assumption may not always be correct, especially when one condition results in overexpression. We present a new method (NormQ) to normalize the RNASeq library size, using the relative proportion observed from RT-qPCR of selected marker genes. The method was compared against the popular median-of-ratios method, using simulated and real-datasets. NormQ produced more matches to differentially expressed genes in the simulated dataset and more distribution profile matches for both simulated and real datasets.
Keywords: DESeq; Median-of-ratios; Normalization; RNASeq; TOMOSeq; Transcriptomics.
© 2020 The Author(s).
Figures

References
-
- Evans C, Hardin J, Stoebel D. Selecting between-sample RNA-Seq normalization methods from the perspective of their assumptions 2016:1–32. https://doi.org/10.1093/bib/bbx008. - PMC - PubMed
-
- Bullard J.H., Purdom E., Hansen K.D., Dudoit S. Evaluation of statistical methods for normalization and differential expression in mRNA-Seq experiments evaluation of statistical methods for normalization and differential expression in mRNA-Seq experiments. UC Berkeley Div Biostat Pap Ser. 2009;11:94. doi: 10.1186/1471-2105-11-94. - DOI - PMC - PubMed
LinkOut - more resources
Full Text Sources
Molecular Biology Databases