

Bi-allelic missense disease-causing variants in RPL3L associate neonatal dilated cardiomyopathy with muscle-specific ribosome biogenesis
- PMID: 32514796
- PMCID: PMC7519902
- DOI: 10.1007/s00439-020-02188-6
Bi-allelic missense disease-causing variants in RPL3L associate neonatal dilated cardiomyopathy with muscle-specific ribosome biogenesis
Abstract
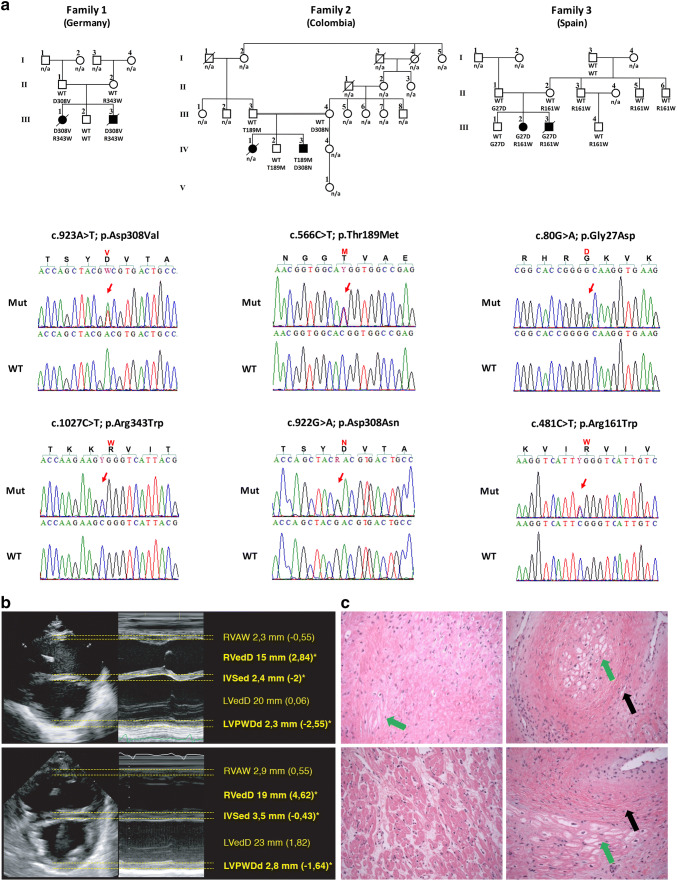
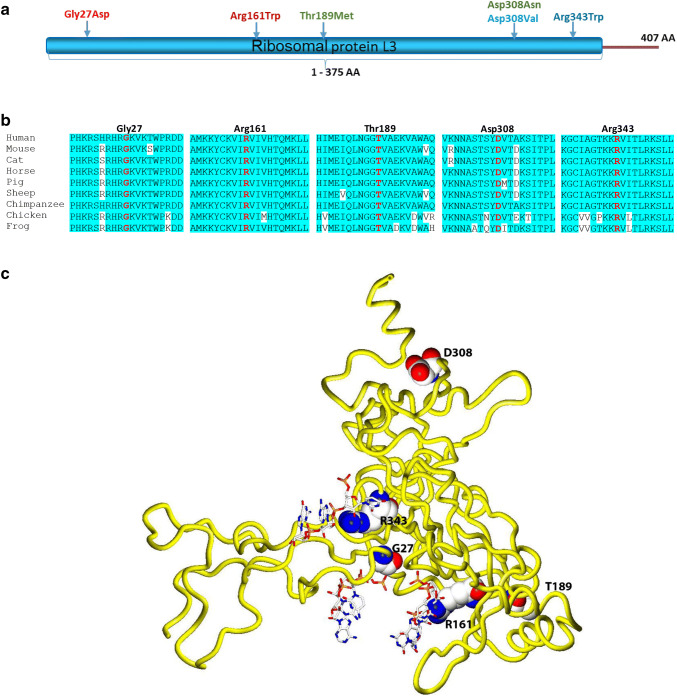
Dilated cardiomyopathy (DCM) belongs to the most frequent forms of cardiomyopathy mainly characterized by cardiac dilatation and reduced systolic function. Although most cases of DCM are classified as sporadic, 20-30% of cases show a heritable pattern. Familial forms of DCM are genetically heterogeneous, and mutations in several genes have been identified that most commonly play a role in cytoskeleton and sarcomere-associated processes. Still, a large number of familial cases remain unsolved. Here, we report five individuals from three independent families who presented with severe dilated cardiomyopathy during the neonatal period. Using whole-exome sequencing (WES), we identified causative, compound heterozygous missense variants in RPL3L (ribosomal protein L3-like) in all the affected individuals. The identified variants co-segregated with the disease in each of the three families and were absent or very rare in the human population, in line with an autosomal recessive inheritance pattern. They are located within the conserved RPL3 domain of the protein and were classified as deleterious by several in silico prediction software applications. RPL3L is one of the four non-canonical riboprotein genes and it encodes the 60S ribosomal protein L3-like protein that is highly expressed only in cardiac and skeletal muscle. Three-dimensional homology modeling and in silico analysis of the affected residues in RPL3L indicate that the identified changes specifically alter the interaction of RPL3L with the RNA components of the 60S ribosomal subunit and thus destabilize its binding to the 60S subunit. In conclusion, we report that bi-allelic pathogenic variants in RPL3L are causative of an early-onset, severe neonatal form of dilated cardiomyopathy, and we show for the first time that cytoplasmic ribosomal proteins are involved in the pathogenesis of non-syndromic cardiomyopathies.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
-
- Abdelfatah N, Chen R, Duff HJ, Seifer CM, Buffo I, Huculak C, Clarke S, Clegg R, Jassal DS, Gordon PMK, Ober C, Care4Rare Canada Consortium, et al (2019) Characterization of a unique form of arrhythmic cardiomyopathy caused by recessive mutation in LEMD2. JACC Basic Transl Sci 4:204–221 - PMC - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
