

PRICKLE3 linked to ATPase biogenesis manifested Leber's hereditary optic neuropathy
- PMID: 32516135
- PMCID: PMC7456240
- DOI: 10.1172/JCI134965
PRICKLE3 linked to ATPase biogenesis manifested Leber's hereditary optic neuropathy
Abstract
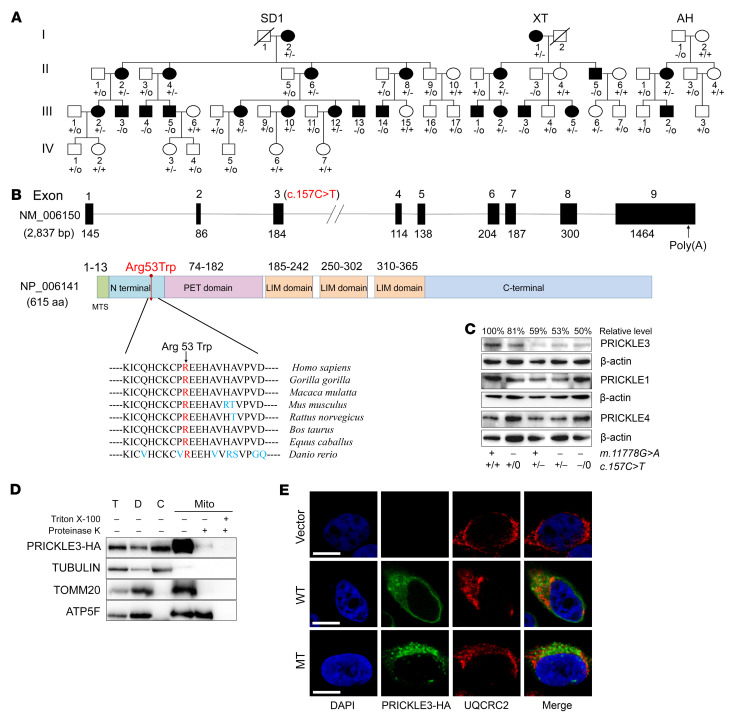
Leber's hereditary optic neuropathy (LHON) is a maternally inherited eye disease. X-linked nuclear modifiers were proposed to modify the phenotypic manifestation of LHON-associated mitochondrial DNA (mtDNA) mutations. By whole-exome sequencing, we identified the X-linked LHON modifier (c.157C>T, p.Arg53Trp) in PRICKLE3 encoding a mitochondrial protein linked to biogenesis of ATPase in 3 Chinese families. All affected individuals carried both ND4 11778G>A and p.Arg53Trp mutations, while subjects bearing only a single mutation exhibited normal vision. The cells carrying the p.Arg53Trp mutation exhibited defective assembly, stability, and function of ATP synthase, verified by PRICKLE3-knockdown cells. Coimmunoprecipitation indicated the direct interaction of PRICKLE3 with ATP synthase via ATP8. Strikingly, cells bearing both p.Arg53Trp and m.11778G>A mutations displayed greater mitochondrial dysfunction than those carrying only a single mutation. This finding indicated that the p.Arg53Trp mutation acted in synergy with the m.11778G>A mutation and deteriorated mitochondrial dysfunctions necessary for the expression of LHON. Furthermore, we demonstrated that Prickle3-deficient mice exhibited pronounced ATPase deficiencies. Prickle3-knockout mice recapitulated LHON phenotypes with retinal deficiencies, including degeneration of retinal ganglion cells and abnormal vasculature. Our findings provided new insights into the pathophysiology of LHON that were manifested by interaction between mtDNA mutations and X-linked nuclear modifiers.
Keywords: Genetic diseases; Genetics; Mitochondria; Molecular pathology; Ophthalmology.
Conflict of interest statement
Figures




References
-
- Newman NJ. Hereditary optic neuropathies: from the mitochondria to the optic nerve. Am J Ophthalmol. 2005;140(3):517–523. - PubMed
-
- Carelli V, La Morgia C, Valentino ML, Barboni P, Ross-Cisneros FN, Sadun AA. Retinal ganglion cell neurodegeneration in mitochondrial inherited disorders. Biochim Biophys Acta. 2009;1787(5):518–528. - PubMed
