

The yeast exoribonuclease Xrn1 and associated factors modulate RNA polymerase II processivity in 5' and 3' gene regions
- PMID: 32518159
- PMCID: PMC7450105
- DOI: 10.1074/jbc.RA120.013426
The yeast exoribonuclease Xrn1 and associated factors modulate RNA polymerase II processivity in 5' and 3' gene regions
Abstract
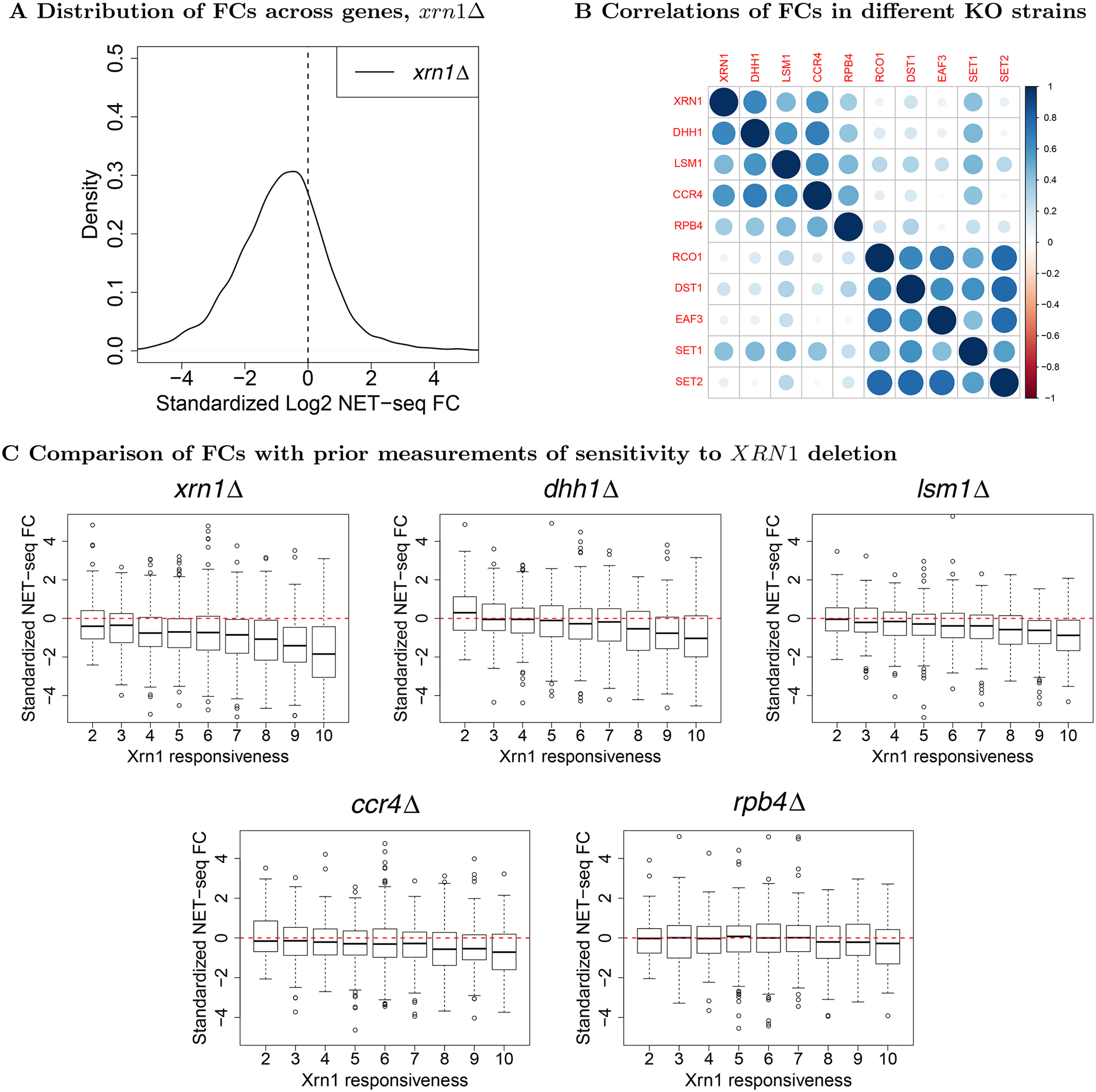
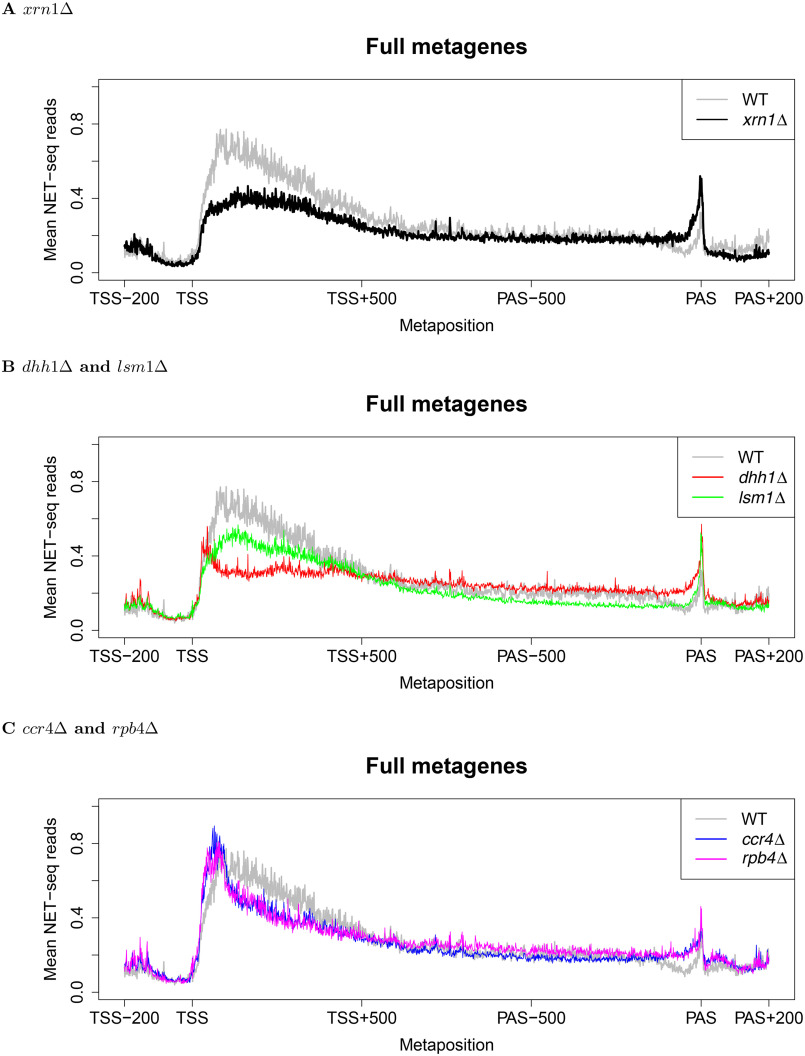
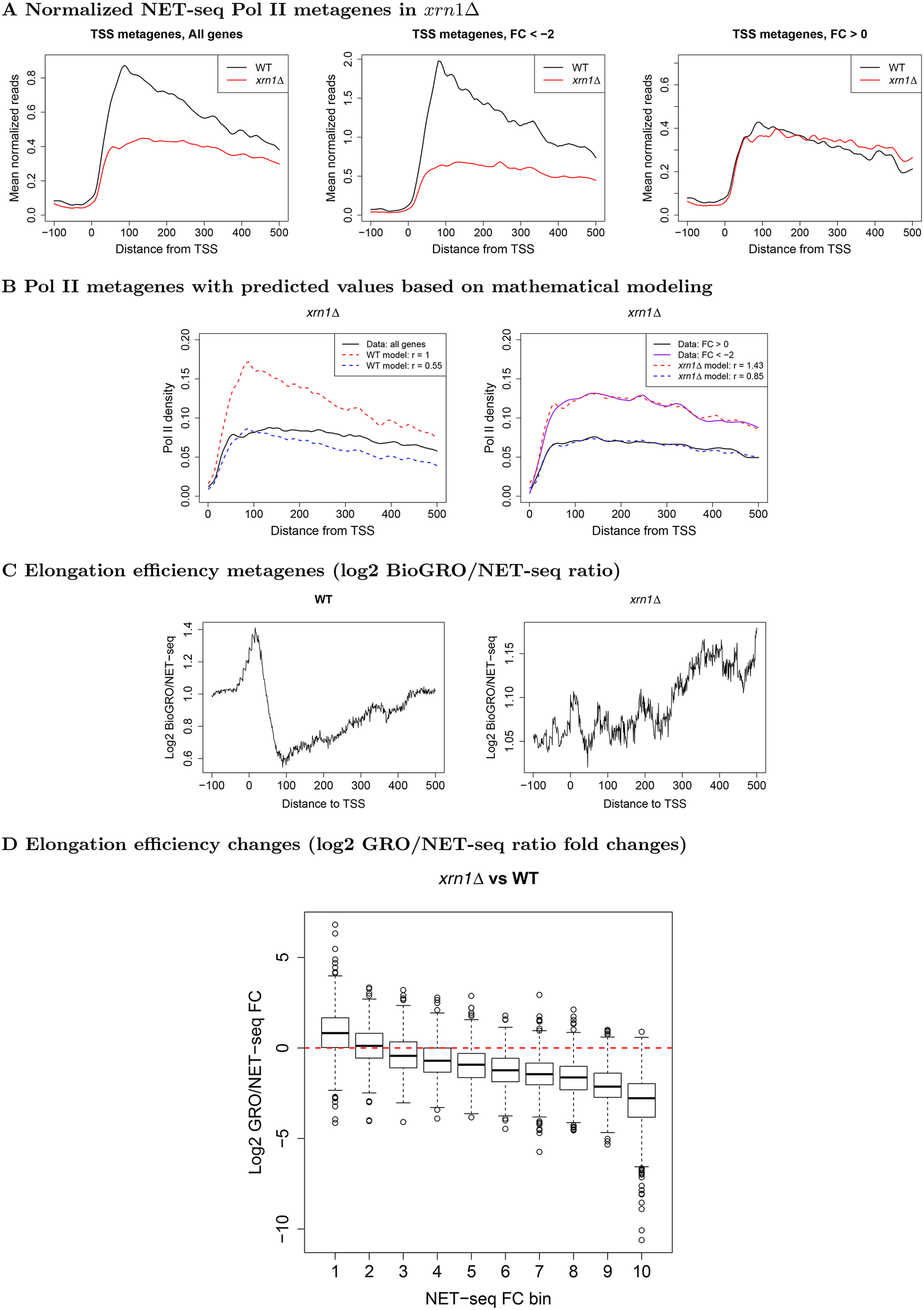
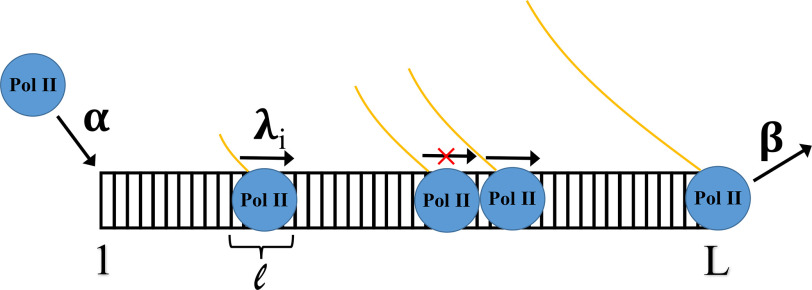
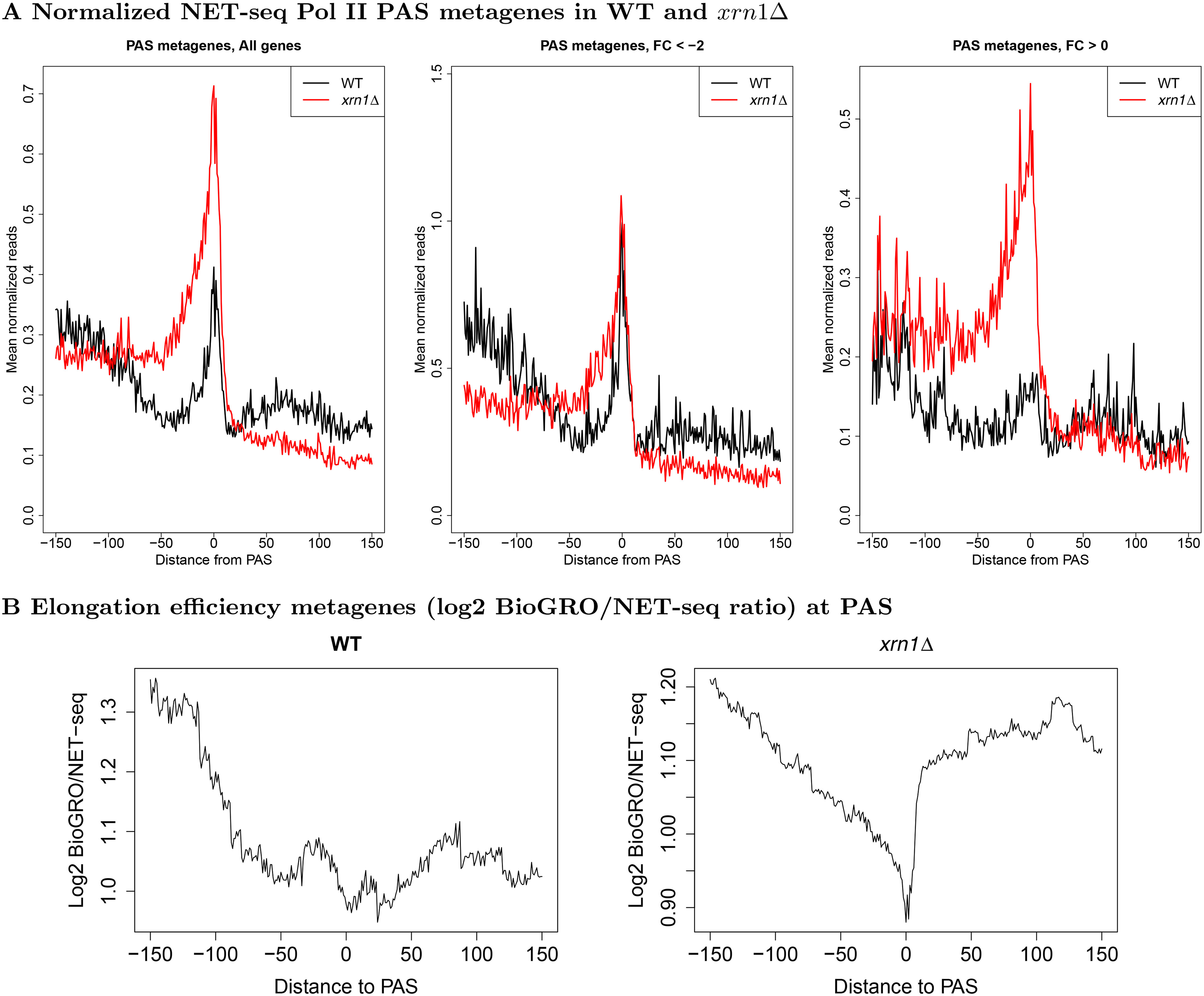
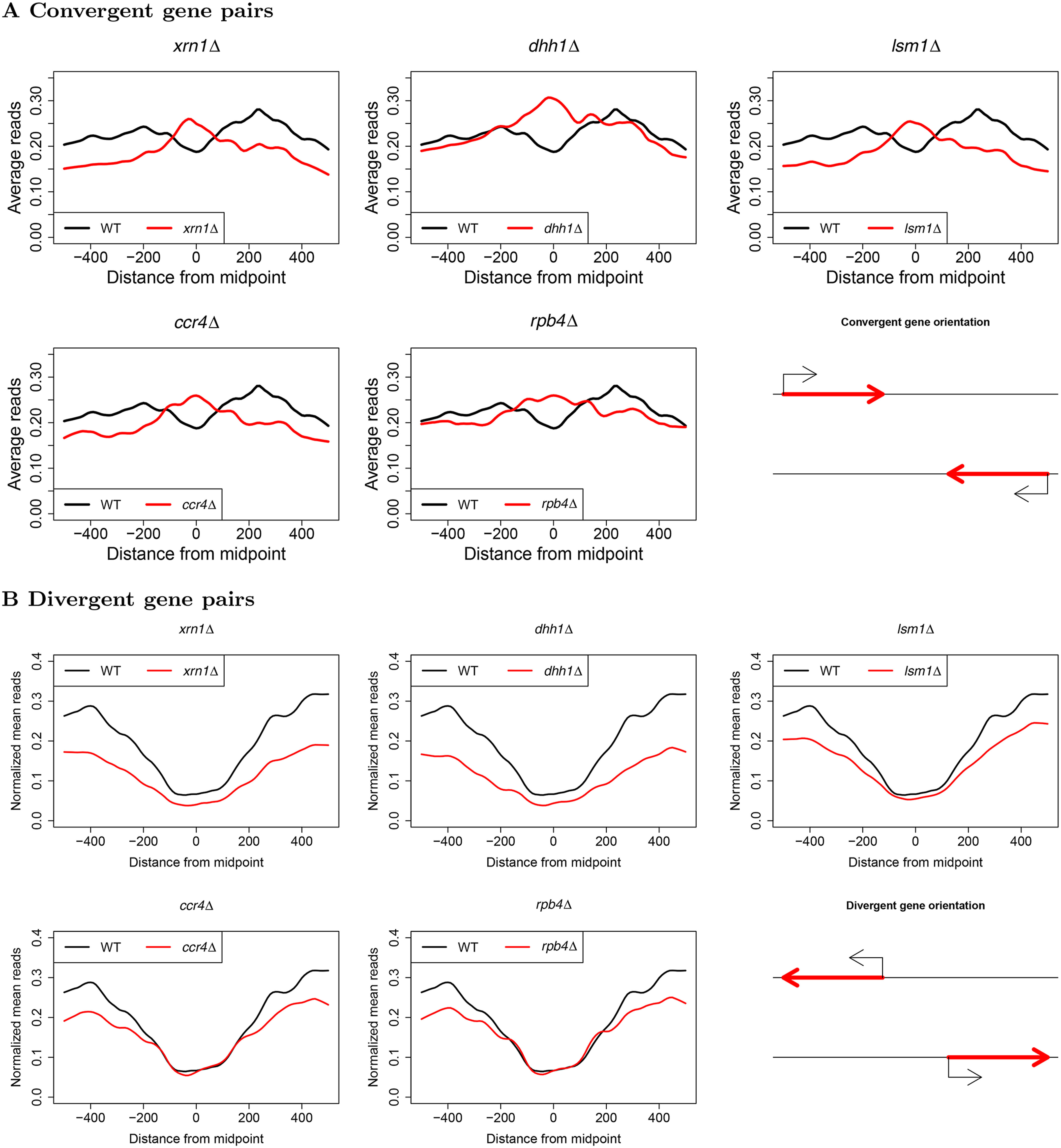
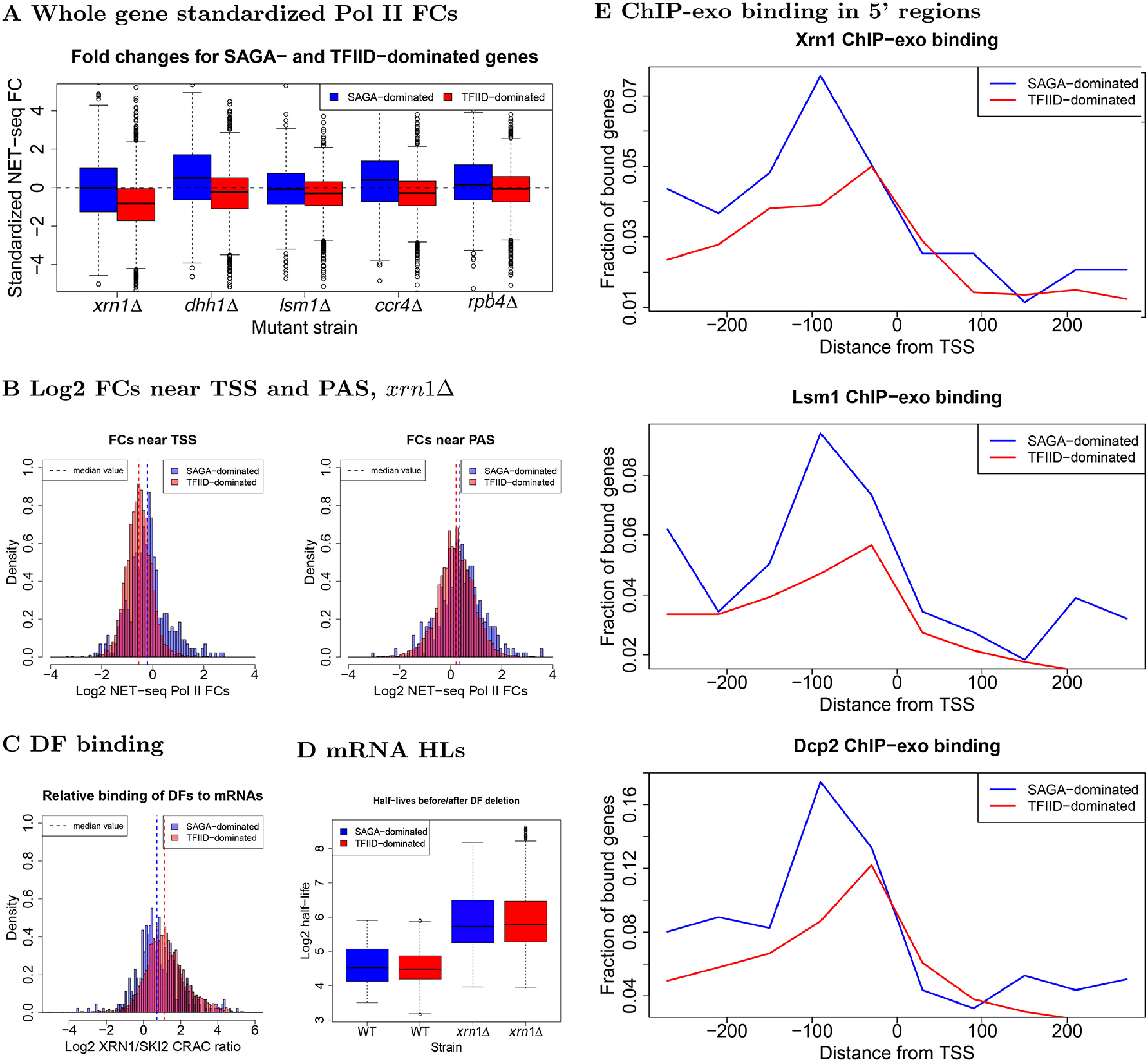
mRNA levels are determined by the balance between mRNA synthesis and decay. Protein factors that mediate both processes, including the 5'-3' exonuclease Xrn1, are responsible for a cross-talk between the two processes that buffers steady-state mRNA levels. However, the roles of these proteins in transcription remain elusive and controversial. Applying native elongating transcript sequencing (NET-seq) to yeast cells, we show that Xrn1 functions mainly as a transcriptional activator and that its disruption manifests as a reduction of RNA polymerase II (Pol II) occupancy downstream of transcription start sites. By combining our sequencing data and mathematical modeling of transcription, we found that Xrn1 modulates transcription initiation and elongation of its target genes. Furthermore, Pol II occupancy markedly increased near cleavage and polyadenylation sites in xrn1Δ cells, whereas its activity decreased, a characteristic feature of backtracked Pol II. We also provide indirect evidence that Xrn1 is involved in transcription termination downstream of polyadenylation sites. We noted that two additional decay factors, Dhh1 and Lsm1, seem to function similarly to Xrn1 in transcription, perhaps as a complex, and that the decay factors Ccr4 and Rpb4 also perturb transcription in other ways. Interestingly, the decay factors could differentiate between SAGA- and TFIID-dominated promoters. These two classes of genes responded differently to XRN1 deletion in mRNA synthesis and were differentially regulated by mRNA decay pathways, raising the possibility that one distinction between these two gene classes lies in the mechanisms that balance mRNA synthesis with mRNA decay.
Keywords: Xrn1; gene regulation; mRNA buffering; mRNA decay; native elongating transcript sequencing (NET-seq); transcription factor; transcription regulation; transcriptional profiling; transcriptomics; yeast.
© 2020 Fischer et al.
Conflict of interest statement
Conflict of interest—The authors declare no conflicts of interest.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
