

Unique and Shared Roles for Histone H3K36 Methylation States in Transcription Regulation Functions
- PMID: 32521276
- PMCID: PMC7334899
- DOI: 10.1016/j.celrep.2020.107751
Unique and Shared Roles for Histone H3K36 Methylation States in Transcription Regulation Functions
Abstract
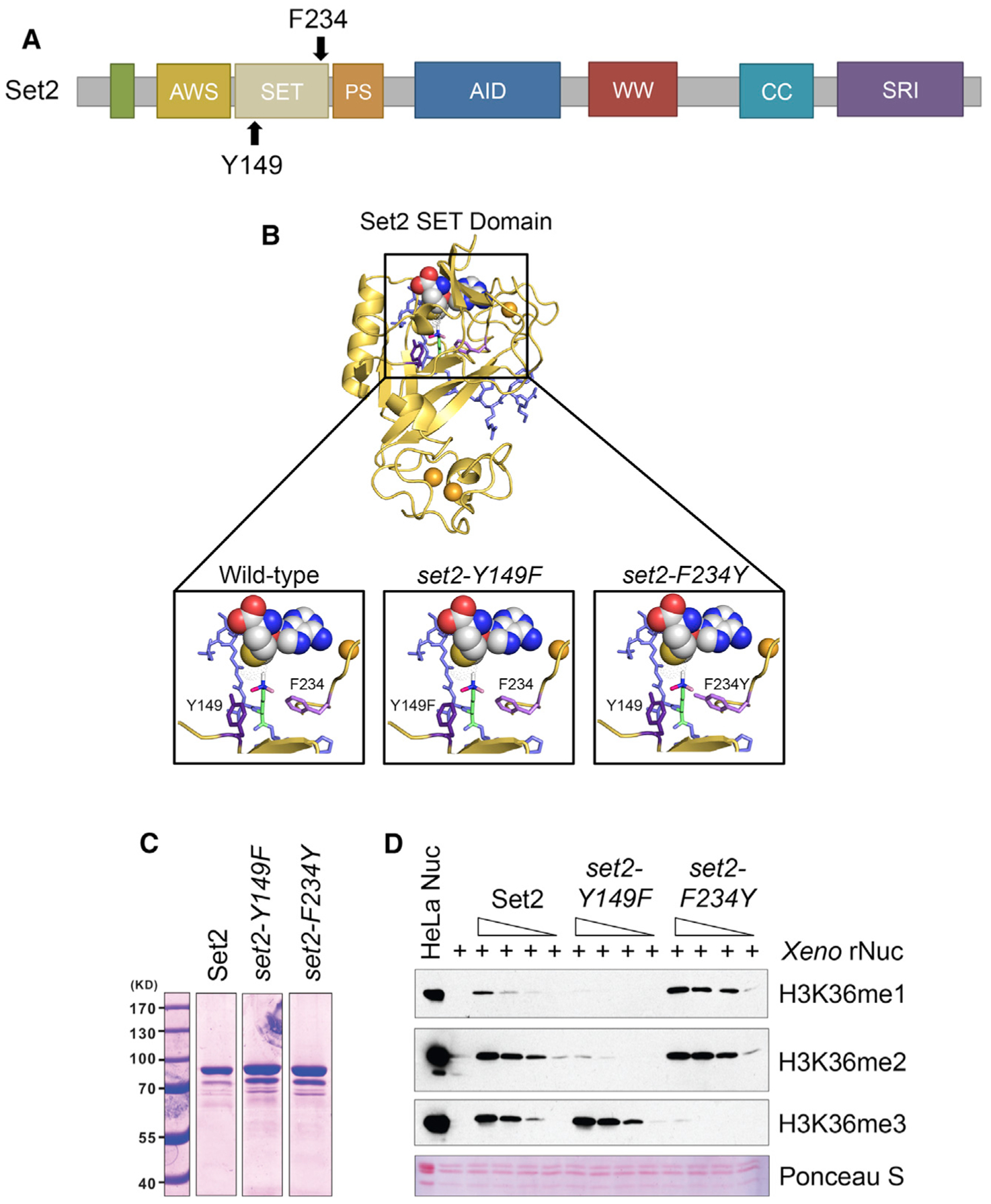
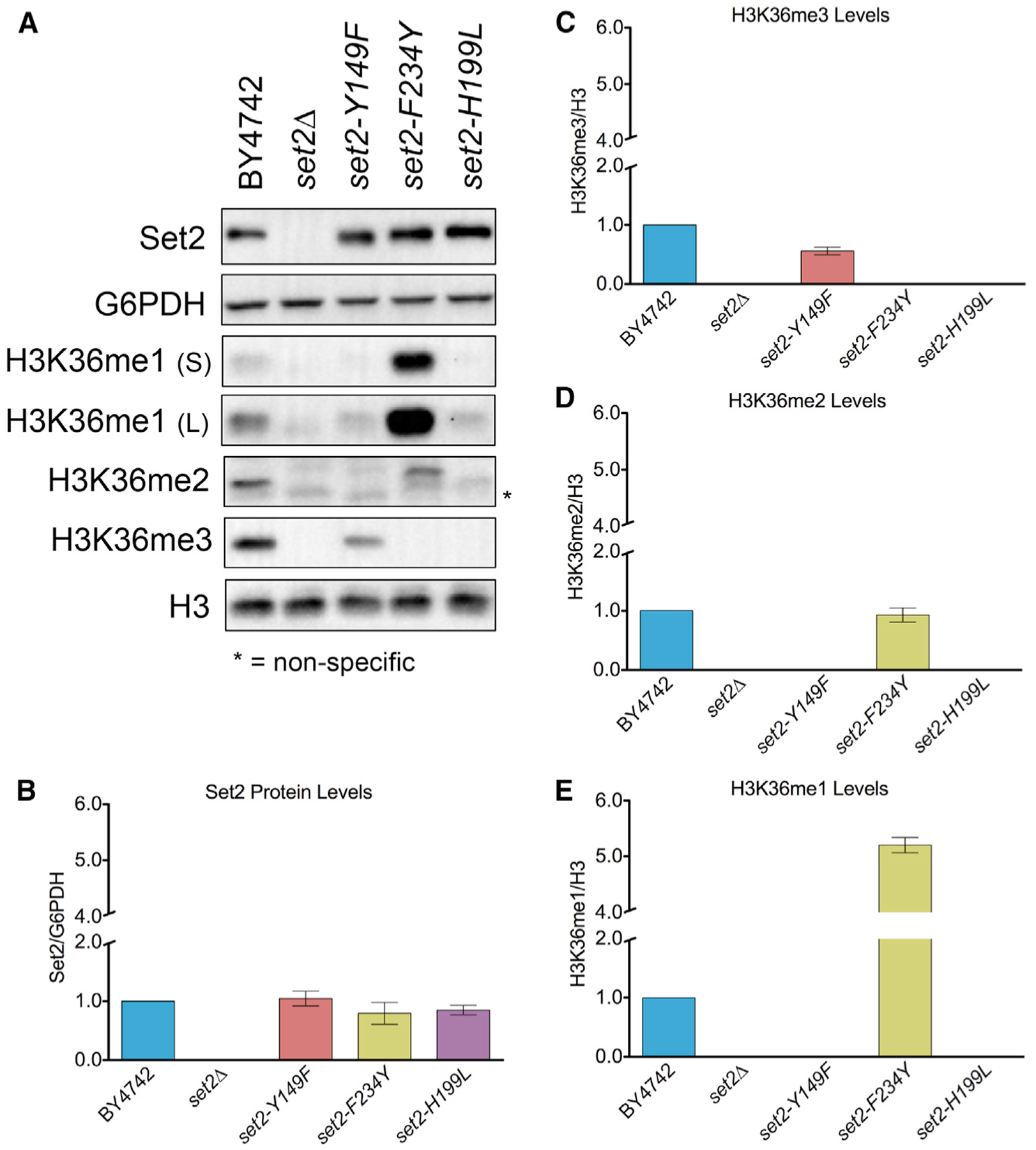
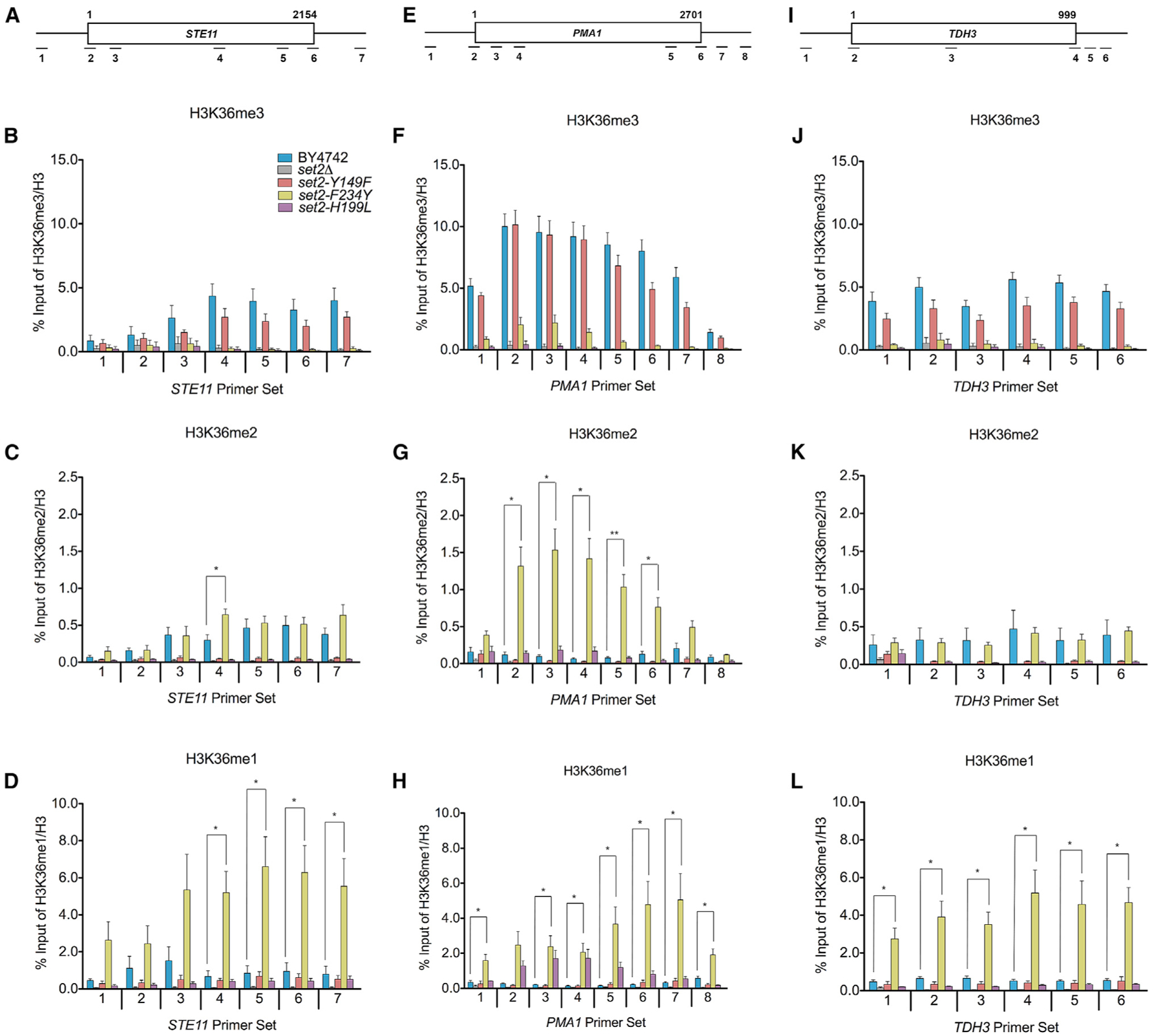
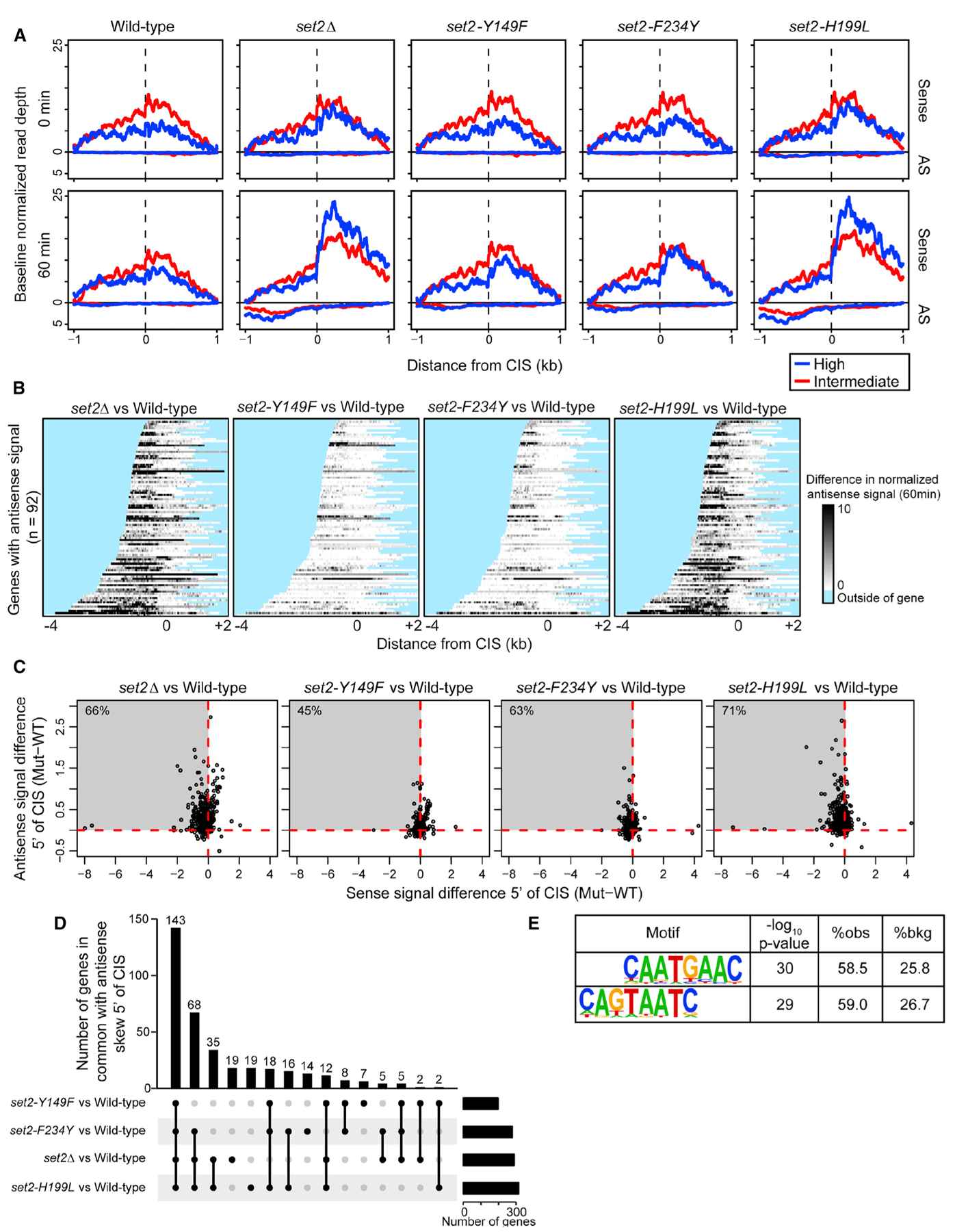
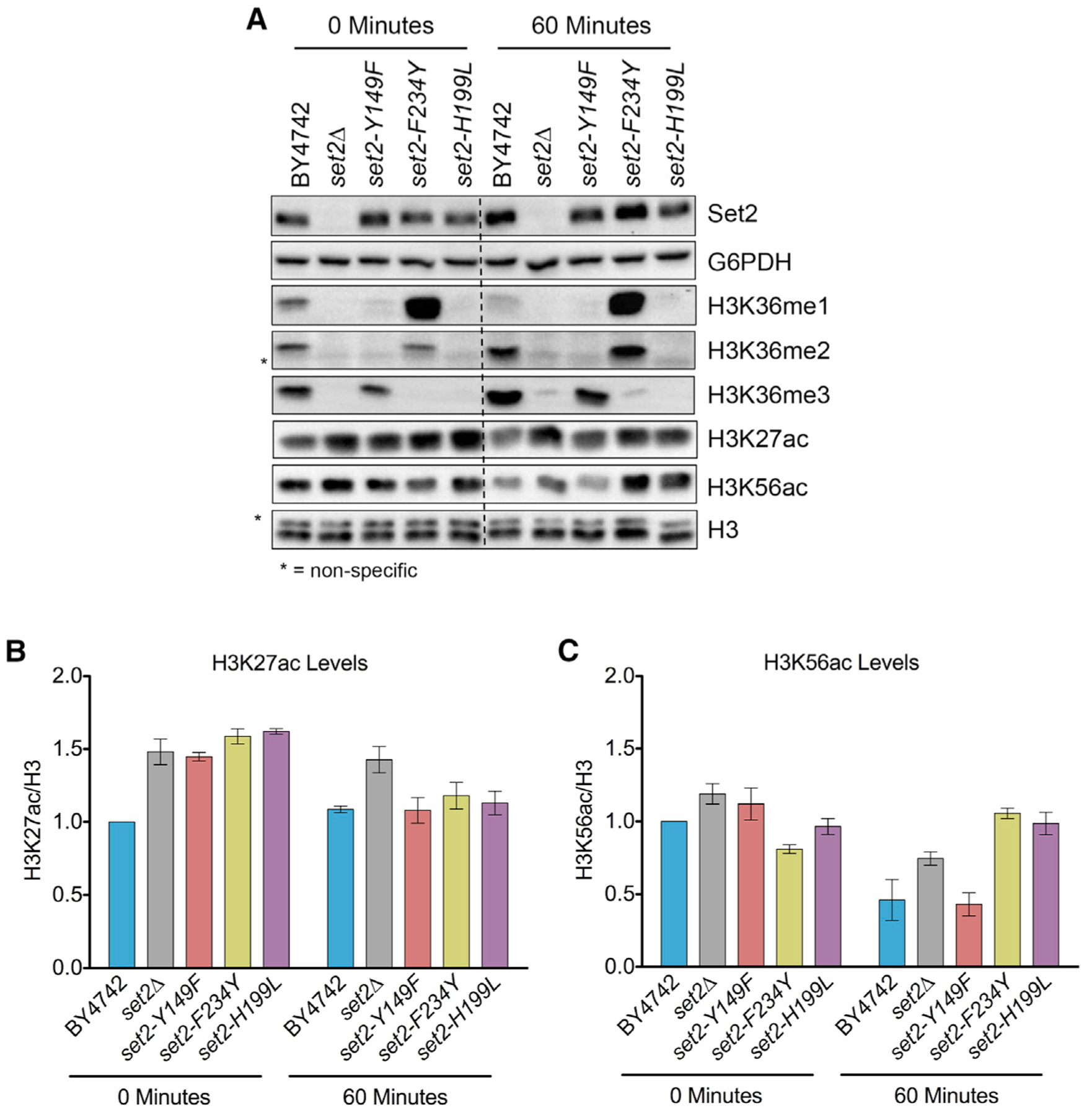
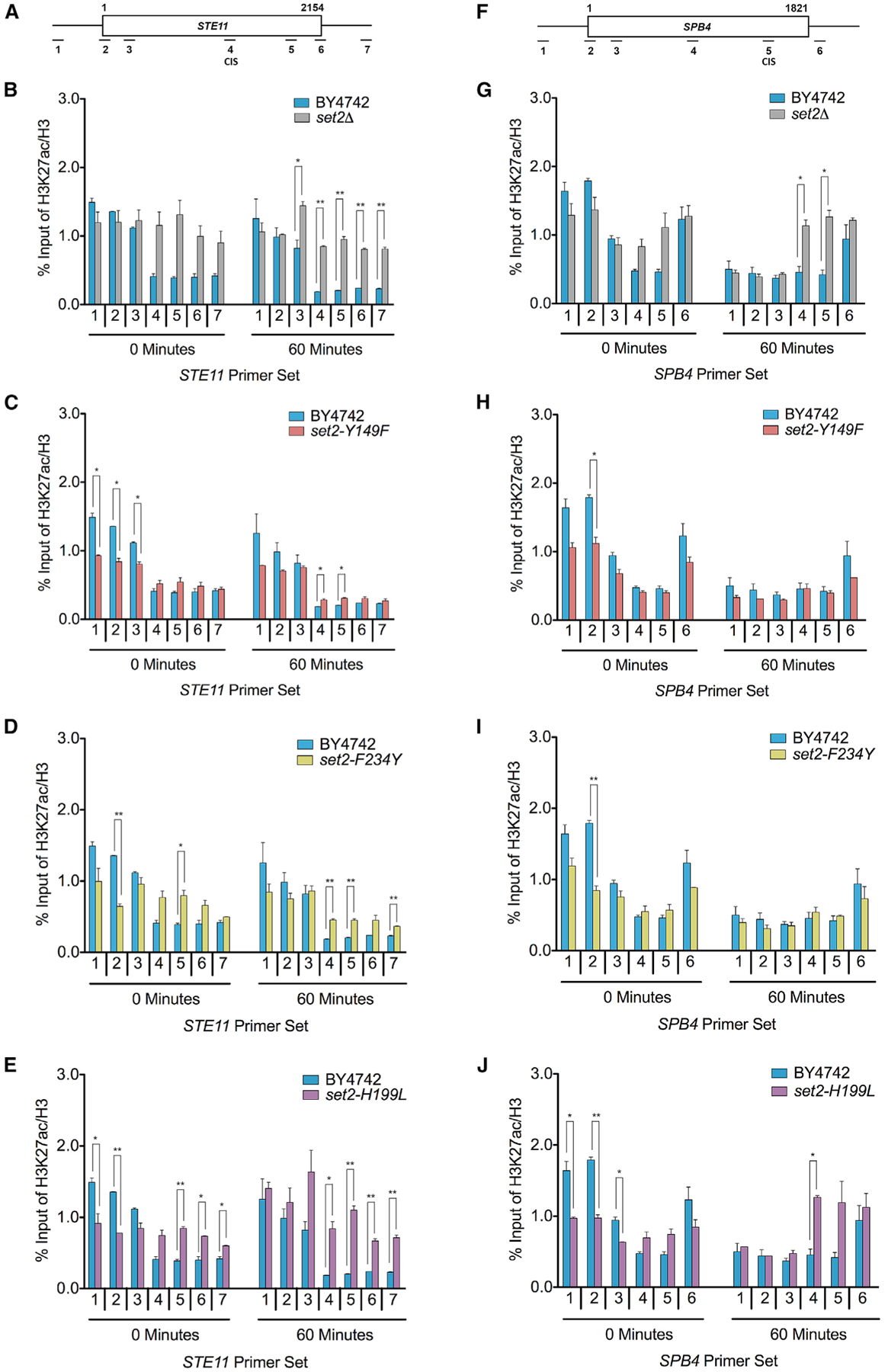
Set2 co-transcriptionally methylates lysine 36 of histone H3 (H3K36), producing mono-, di-, and trimethylation (H3K36me1/2/3). These modifications recruit or repel chromatin effector proteins important for transcriptional fidelity, mRNA splicing, and DNA repair. However, it was not known whether the different methylation states of H3K36 have distinct biological functions. Here, we use engineered forms of Set2 that produce different lysine methylation states to identify unique and shared functions for H3K36 modifications. Although H3K36me1/2 and H3K36me3 are functionally redundant in many SET2 deletion phenotypes, we found that H3K36me3 has a unique function related to Bur1 kinase activity and FACT (facilitates chromatin transcription) complex function. Further, during nutrient stress, either H3K36me1/2 or H3K36me3 represses high levels of histone acetylation and cryptic transcription that arises from within genes. Our findings uncover the potential for the regulation of diverse chromatin functions by different H3K36 methylation states.
Keywords: H3K36 methylation; RNA Polymerase II; Set2; chromatin; cryptic transcription; epigenetics; histone; nutrient stress; transcriptional regulation.
Copyright © 2020 The Author(s). Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of Interests B.D.S. acknowledges he is a co-founder and scientific advisory board member of EpiCypher, Inc.
Figures

References
-
- Basehoar AD, Zanton SJ, and Pugh BF (2004). Identification and distinct regulation of yeast TATA box-containing genes. Cell 116, 699–709. - PubMed
-
- Carrozza MJ, Li B, Florens L, Suganuma T, Swanson SK, Lee KK, Shia WJ, Anderson S, Yates J, Washburn MP, and Workman JL (2005). Histone H3 methylation by Set2 directs deacetylation of coding regions by Rpd3S to suppress spurious intragenic transcription. Cell 123, 581–592. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
