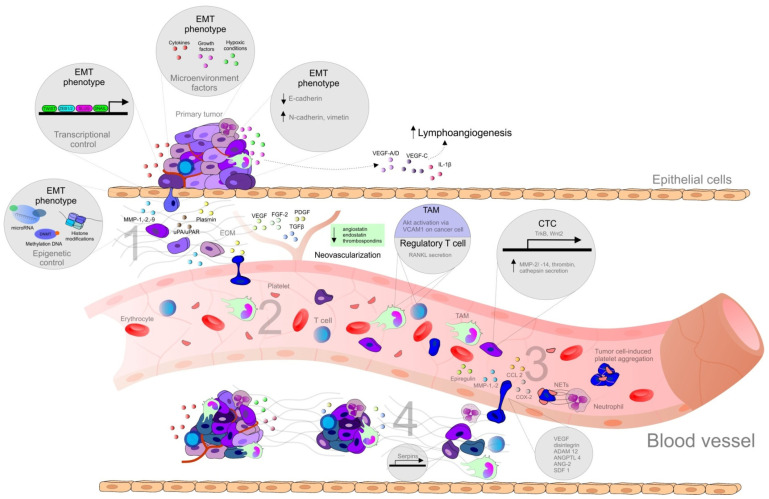
Figure 1
Steps of the metastatic spread from the primary tumor. Abbreviations: ADAM12, metalloproteinase domain-containing protein 12; ANG-2, angiotensin II; ANGPTL4, angiopoietin-like 4; CCL2, CC-chemokine ligand 2; COX-2, cyclooxygenase-2; CTCs, circulating tumor cells; DNMT, DNA methyltransferase; DTCs, disseminated tumor cells; ECM, extracellular matrix; EMT, epithelial-mesenchymal transition; FGF, fibroblast growth factor; IL, interleukin; MAMs, metastasis-associated macrophages; MMPs, matrix metalloproteinase; NET, neutrophil extracellular traps; PDGF, platelet-derived growth factor; RANKL, regulatory T cells producing receptor activator of nuclear factor-κB ligand; SDF1, stromal cell-derived factor 1; Snail, Slug, transcription factors of a snail family; TAM, tumor associated macrophages; TCs, tumor cells; TGFβ, transforming growth factor beta; Twist, basic helix-loop-helix factors; uPA/uPAR, urokinase plasminogen activator/urokinase plasminogen activator receptor; VCAM1, vascular cell adhesion molecule 1; VEGF, vascular endothelial growth factor; ZEB, zinc-finger E-box-binding factors such as the two-handed zinc-finger factors of d-crystallin/E2 box factor (dEF1) family proteins dEF1/ZEB homeobox 1 and Smad-interacting protein 1/ZEB2. Explanatory notes: 1. Local invasion: an initiation and maintenance of cancer invasion is facilitated by the regulation of cytoskeletal dynamics in cancer cells and the cell-ECM and cell-cell junctions turnover [14]. EMT that allows cancer cells to accomplish migration and invasion [13] is induced by various stimuli, such as hypoxia, cytokines, and growth factors [15]. EMT is regulated by transcriptional factors (Twist, Snail, Slug, ZEB1, ZEB2) or by epigenetic regulation [15,16]. EMT phenotype is represented by downregulation of E-cadherin and upregulation of N-cadherin (cadherin switch), and vimentin [17]. Remodeling of ECM contributes to cancer progression (clustering of integrins and other receptors → activation of intracellular kinase signaling pathways altering EMT, cancer migration, and invasion) [14]. The detachment from the primary lesions, EMT, migration, and invasion through the basement membrane is followed by degradation of ECM of cancer cells by MMPs (-1, -2, -9) and uPA/uPAR [13]: uPA-uPAR binding → activated uPA catalyzes the conversion of plasminogen to plasmin → degradation of ECM (direct or indirect via activation of MMPs) [18]. Tumor cells escape from immune response and establish a tumor-supportive environment (pre-metastatic niche) in the site of future metastasis [19]. 2. Intravasation and survival in the circulation: the active entry of tumor cells into the circulation is promoted by MMPs or uPA/uPAR [13]. Metastatic process is associated with tumor neovascularization (the secretion of pro-angiogenic stimuli-VEGF, FGF-2, ILs, PlGF, TGF-β, PDGF, angiopoietins etc., and downregulation of anti-angiogenic factors, such as endostatins, angiostatin or thrombospondins) [20], and lymphangiogenesis (VEGF-A/D, VEGF-C, IL-1β, FGF, ECM components, activation of the sympathetic nervous system by chronic stress) [14]. CTCs (overexpressing TrkB or Wnt2) facilitate the avoidance of death stress for epithelial cells from anchorage detachment (anoikis). Other mechanisms allowing the survival of cancer cells in the circulation include the protection against physical shear forces and predation of natural killer cells (secretion of substances, such as thrombin, cathepsin B, MMP-2/-14), the formation of clusters of CTCs and platelets also known as tumor cell-induced platelet aggregation [13,21], and survival signals of TAMs, which activate AKT signaling through vascular cell adhesion molecule 1 (VCAM1) on cancer cells, and regulatory T cells producing receptor activator of nuclear factor-κB ligand (RANKL) [19]. 3. Arrest at distant sites and extravasation: process of CTCs leaving the blood flow (extravasation), [13] facilitating the disruption of vascular junctions and invasion of cancer cells into distant organs, is promoted by epiregulin (EREG), MMPs, COX-2, fascin, and differences in the vascular features of distant organs and primary tumor require additional genes (e.g., ANGPTL4) [21]. Metastatic colonization occurs only at certain sites [14] and is facilitated by release of organ-specific chemokines and the expression of appropriate receptors on the surface of the tumor [13], stimulation of neutrophils by cancer cells → neutrophil extracellular NETs supporting metastatic colonization of distant sites. Vascular permeability is modulated by CCL2, VEGF, disintegrin, ADAM12, epiregulin, COX-2, MMP-1, MMP-2, ANG-2, and SDF1 [14]. Cancer cells trapped in an emboli (metastatic site) produce CCL2 to recruit inflammatory monocytes toward metastatic regions, which differentiate into MAMs secreting VEGF [19]. 4. Micrometastasis formation and metastatic colonization: a challenge for just arriving cancer cells include the differences in stromal components, tissue organization, matrix composition, and cytokine environment [21]. Tissue-specific events are required for the survival in sites of various organs [14]. DTCs facilitating the expression of markers of non-cancerous resident cells or mimicking them represent a possible adaptation for survival and allow secondary organ colonization, as was demonstrated in DTCs expressing serpins, that are typically produced by neurons to protect against plasminogen activator-mediated cell death. Moreover, arriving cancer cells are able to subvert resident stromal cells in order to remodel new environments [14].