

mTADA is a framework for identifying risk genes from de novo mutations in multiple traits
- PMID: 32522981
- PMCID: PMC7287090
- DOI: 10.1038/s41467-020-16487-z
mTADA is a framework for identifying risk genes from de novo mutations in multiple traits
Abstract
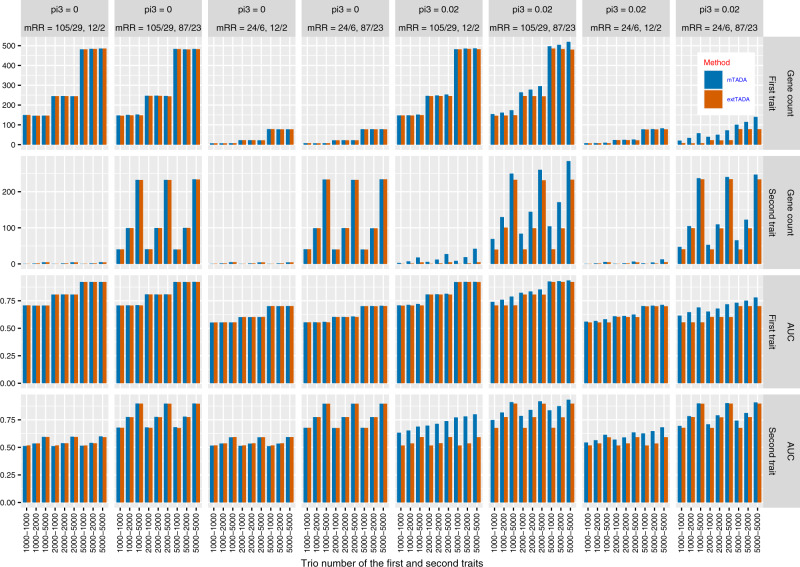
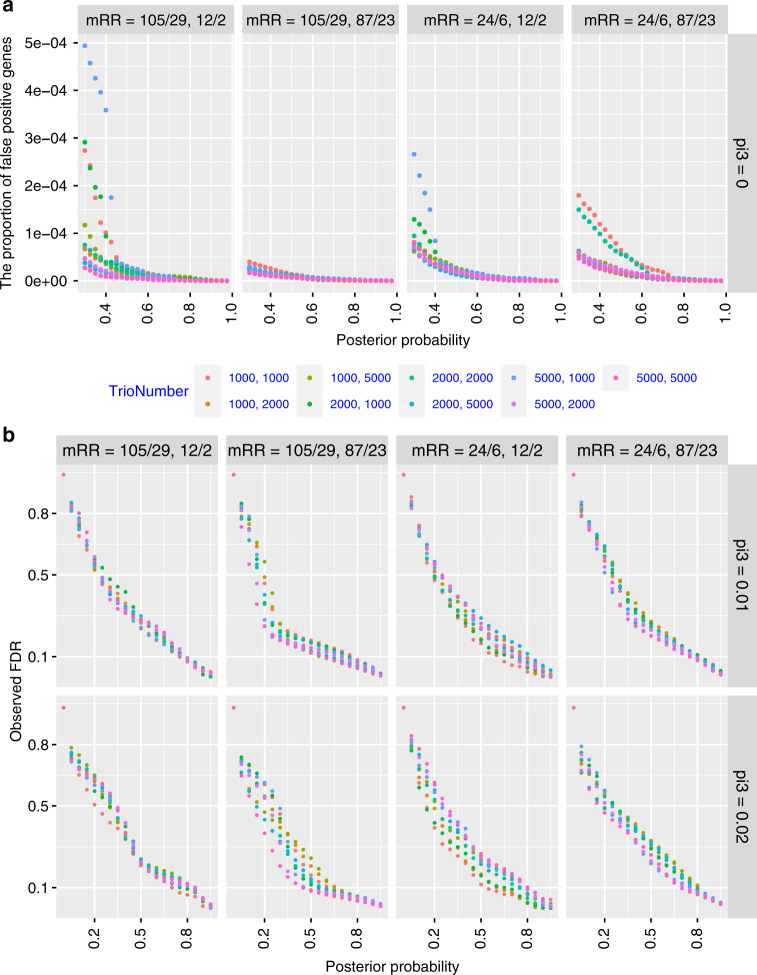
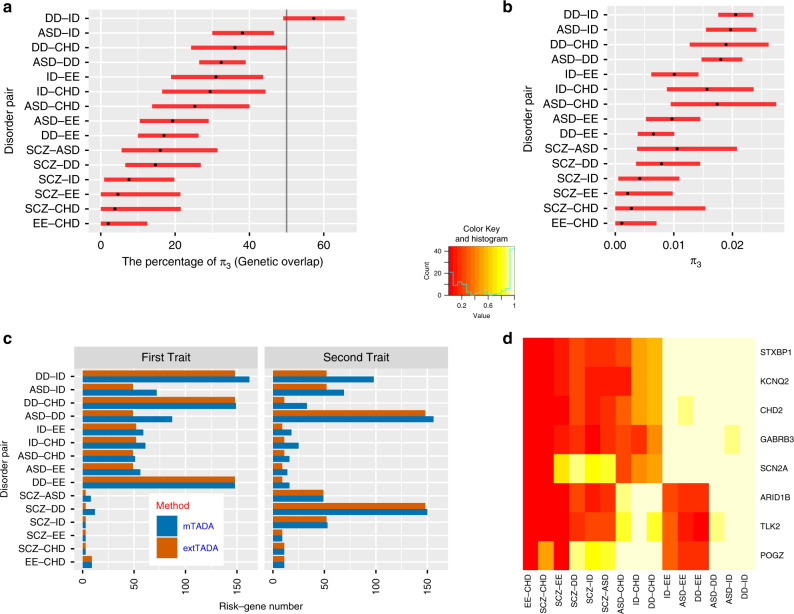
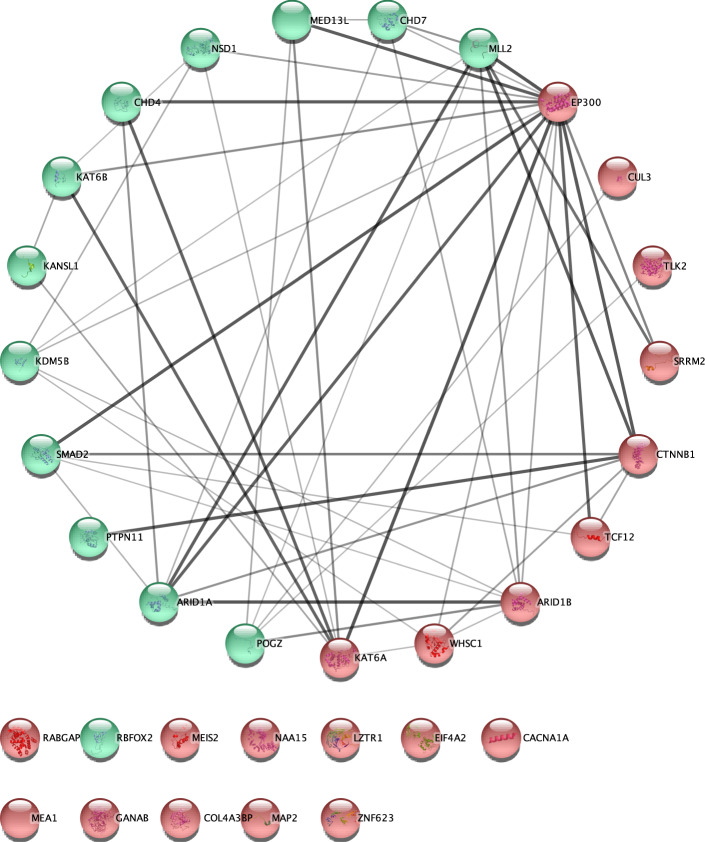
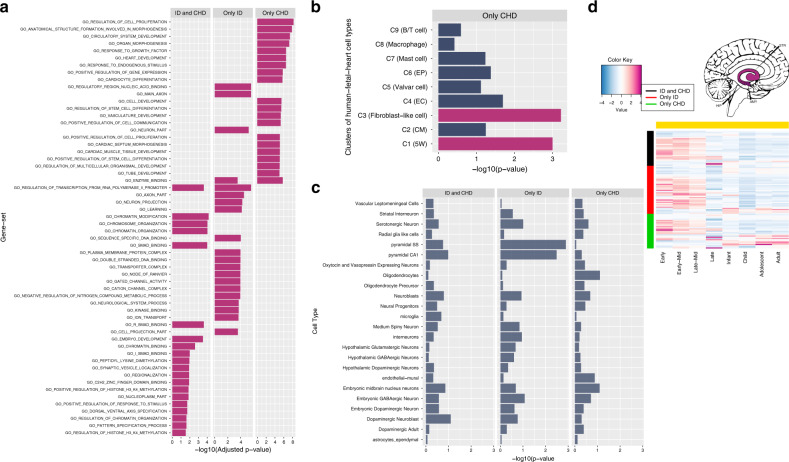
Joint analysis of multiple traits can result in the identification of associations not found through the analysis of each trait in isolation. Studies of neuropsychiatric disorders and congenital heart disease (CHD) which use de novo mutations (DNMs) from parent-offspring trios have reported multiple putatively causal genes. However, a joint analysis method designed to integrate DNMs from multiple studies has yet to be implemented. We here introduce multiple-trait TADA (mTADA) which jointly analyzes two traits using DNMs from non-overlapping family samples. We first demonstrate that mTADA is able to leverage genetic overlaps to increase the statistical power of risk-gene identification. We then apply mTADA to large datasets of >13,000 trios for five neuropsychiatric disorders and CHD. We report additional risk genes for schizophrenia, epileptic encephalopathies and CHD. We outline some shared and specific biological information of intellectual disability and CHD by conducting systems biology analyses of genes prioritized by mTADA.
Conflict of interest statement
P.F.S. reports the following potentially competing financial interests: Lundbeck (advisory committee, grant recipient), Pfizer (Scientific Advisory Board), Element Genomics (consultation fee), and Roche (speaker reimbursement). The remaining authors declare that they have no competing interests.
Figures

References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
