

Properties of structural variants and short tandem repeats associated with gene expression and complex traits
- PMID: 32522982
- PMCID: PMC7286898
- DOI: 10.1038/s41467-020-16482-4
Properties of structural variants and short tandem repeats associated with gene expression and complex traits
Abstract
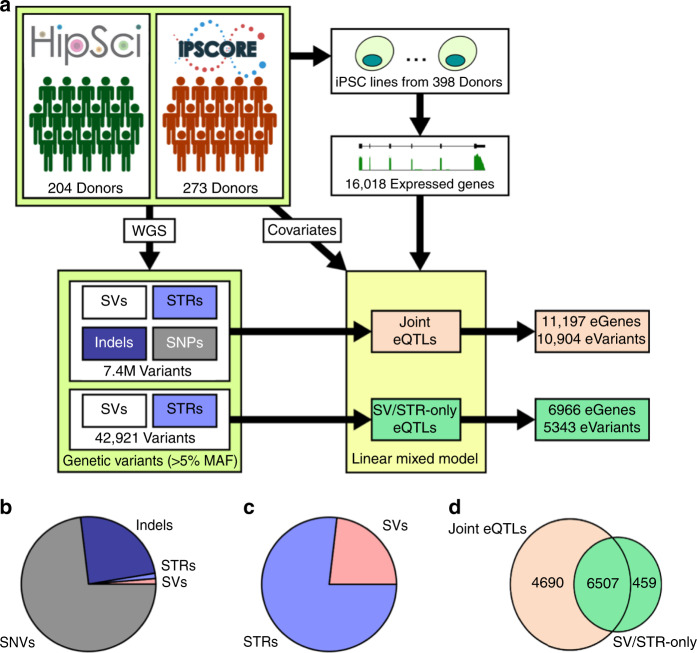
Structural variants (SVs) and short tandem repeats (STRs) comprise a broad group of diverse DNA variants which vastly differ in their sizes and distributions across the genome. Here, we identify genomic features of SV classes and STRs that are associated with gene expression and complex traits, including their locations relative to eGenes, likelihood of being associated with multiple eGenes, associated eGene types (e.g., coding, noncoding, level of evolutionary constraint), effect sizes, linkage disequilibrium with tagging single nucleotide variants used in GWAS, and likelihood of being associated with GWAS traits. We identify a set of high-impact SVs/STRs associated with the expression of three or more eGenes via chromatin loops and show that they are highly enriched for being associated with GWAS traits. Our study provides insights into the genomic properties of structural variant classes and short tandem repeats that are associated with gene expression and human traits.
Conflict of interest statement
The authors declare no competing interests.
Figures





References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
