

Amyotrophic lateral sclerosis: a clinical review
- PMID: 32526057
- PMCID: PMC7540334
- DOI: 10.1111/ene.14393
Amyotrophic lateral sclerosis: a clinical review
Abstract
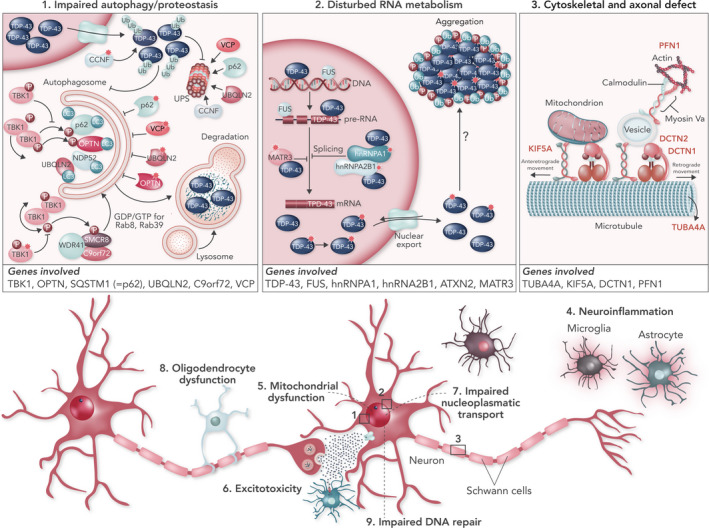
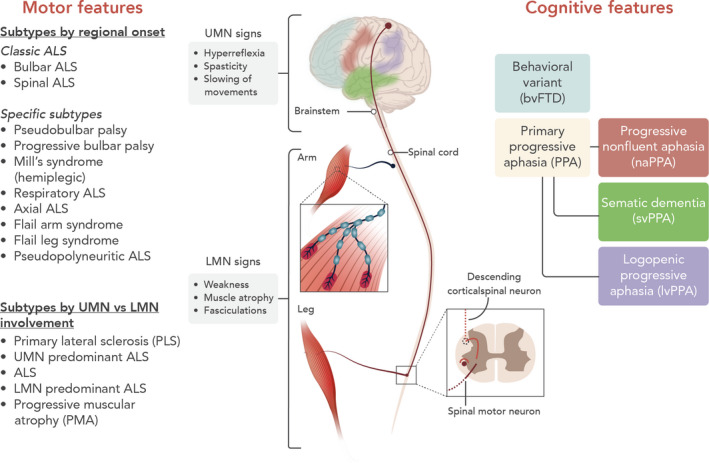
Amyotrophic lateral sclerosis (ALS) is a neurodegenerative disorder affecting primarily the motor system, but in which extra-motor manifestations are increasingly recognized. The loss of upper and lower motor neurons in the motor cortex, the brain stem nuclei and the anterior horn of the spinal cord gives rise to progressive muscle weakness and wasting. ALS often has a focal onset but subsequently spreads to different body regions, where failure of respiratory muscles typically limits survival to 2-5 years after disease onset. In up to 50% of cases, there are extra-motor manifestations such as changes in behaviour, executive dysfunction and language problems. In 10%-15% of patients, these problems are severe enough to meet the clinical criteria of frontotemporal dementia (FTD). In 10% of ALS patients, the family history suggests an autosomal dominant inheritance pattern. The remaining 90% have no affected family members and are classified as sporadic ALS. The causes of ALS appear to be heterogeneous and are only partially understood. To date, more than 20 genes have been associated with ALS. The most common genetic cause is a hexanucleotide repeat expansion in the C9orf72 gene, responsible for 30%-50% of familial ALS and 7% of sporadic ALS. These expansions are also a frequent cause of frontotemporal dementia, emphasizing the molecular overlap between ALS and FTD. To this day there is no cure or effective treatment for ALS and the cornerstone of treatment remains multidisciplinary care, including nutritional and respiratory support and symptom management. In this review, different aspects of ALS are discussed, including epidemiology, aetiology, pathogenesis, clinical features, differential diagnosis, investigations, treatment and future prospects.
Keywords: TDP-43 pathology; amyotrophic lateral sclerosis; sporadic and familial ALS.
© 2020 The Authors. European Journal of Neurology published by John Wiley & Sons Ltd on behalf of European Academy of Neurology.
Conflict of interest statement
The authors declare no financial or other conflicts of interest.
Figures
References
-
- Hardiman O, Al‐Chalabi A, Chio A, et al Amyotrophic lateral sclerosis. Nat Rev Dis Primers 2017; 3: 17085. - PubMed
-
- Brown RH, Al‐Chalabi A. Amyotrophic lateral sclerosis. N Engl J Med 2017; 377: 162–172. - PubMed
-
- van Es MA, Hardiman O, Chio A, et al Amyotrophic lateral sclerosis. Lancet 2017; 390: 2084–2098. - PubMed
-
- Phukan J, Pender NP, Hardiman O. Cognitive impairment in amyotrophic lateral sclerosis. Lancet Neurol 2007; 6: 994–1003. - PubMed
-
- Neary D, Snowden JS, Gustafson L, et al Frontotemporal lobar degeneration: a consensus on clinical diagnostic criteria. Neurology 1998; 51: 1546–1554. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- C1-C14-17-107/KU Leuven/International
- Opening the Future Fund (KU Leuven)/International
- the Fund for Scientific Research Flanders (FWO-Flanders)/International
- the ALS Liga Belgium/International
- Een Hart voor ALS/International
- '/International
- Laeversfonds voor ALS Onderzoek/International
- Valéry Perrier Race against ALS Fund/International
- SAO-FRA 2017/023/the Alzheimer Research Foundation/International
- VIND 135043/the Flemish Government initiated Flanders Impulse Program on Networks for Dementia Research/International
- Flanders Innovation and Enterpreneurship (IWT grants Project MinE and iPSCAF)/International
- the Belgian National Lottery/International
- the Latran Foundation/International
- 755094/the European Union's Horizon 2020 research and innovation programme/International
- the European Union's ERA-Net for Research Programmes on Rare Diseases (INTEGRALS)/International
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
