

Personalized Mapping of Drug Metabolism by the Human Gut Microbiome
- PMID: 32526207
- PMCID: PMC8591631
- DOI: 10.1016/j.cell.2020.05.001
Personalized Mapping of Drug Metabolism by the Human Gut Microbiome
Abstract
The human gut microbiome harbors hundreds of bacterial species with diverse biochemical capabilities. Dozens of drugs have been shown to be metabolized by single isolates from the gut microbiome, but the extent of this phenomenon is rarely explored in the context of microbial communities. Here, we develop a quantitative experimental framework for mapping the ability of the human gut microbiome to metabolize small molecule drugs: Microbiome-Derived Metabolism (MDM)-Screen. Included are a batch culturing system for sustained growth of subject-specific gut microbial communities, an ex vivo drug metabolism screen, and targeted and untargeted functional metagenomic screens to identify microbiome-encoded genes responsible for specific metabolic events. Our framework identifies novel drug-microbiome interactions that vary between individuals and demonstrates how the gut microbiome might be used in drug development and personalized medicine.
Keywords: drug metabolism; drug-microbiome interactions; functional metagenomics; gut microbiome; microbial community; personalized medicine.
Copyright © 2020 Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of Interests M.S.D. is a member of the scientific advisory board of DeepBiome Therapeutics. A patent is being filed by Princeton University for the use of quantitative MDM-Screen to measure inter-individual variability in drug metabolism.
Figures
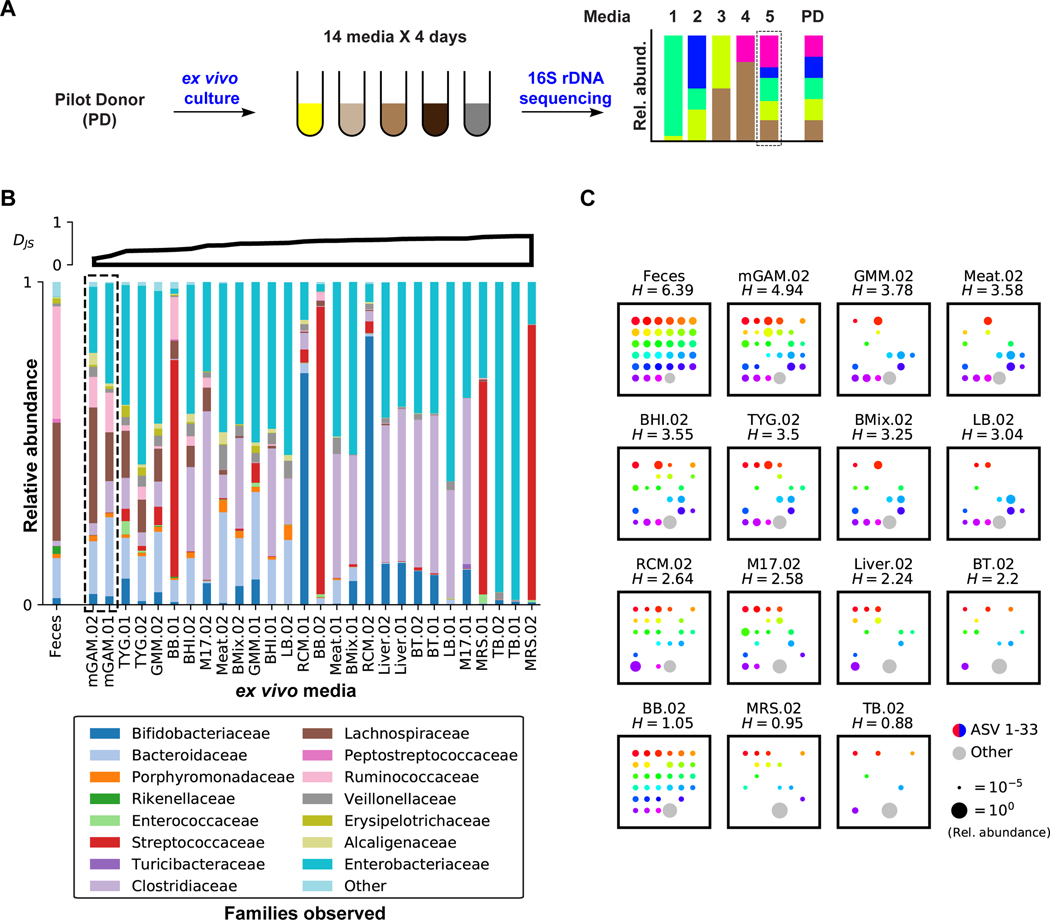
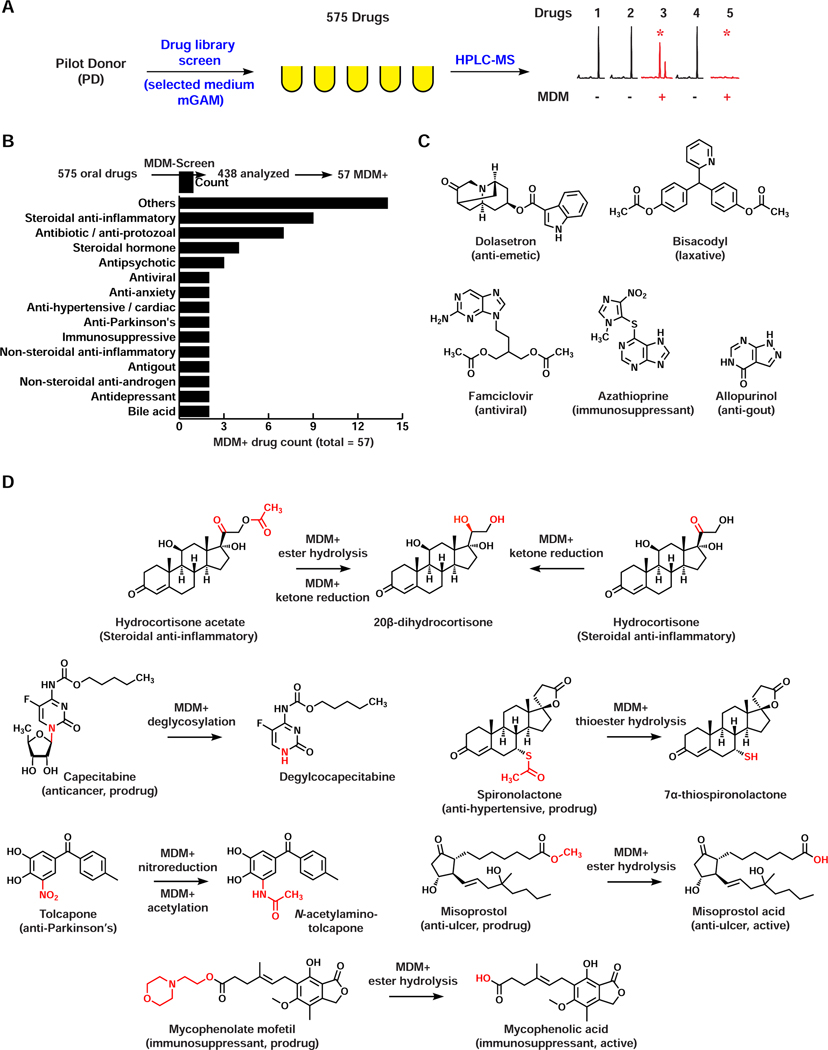
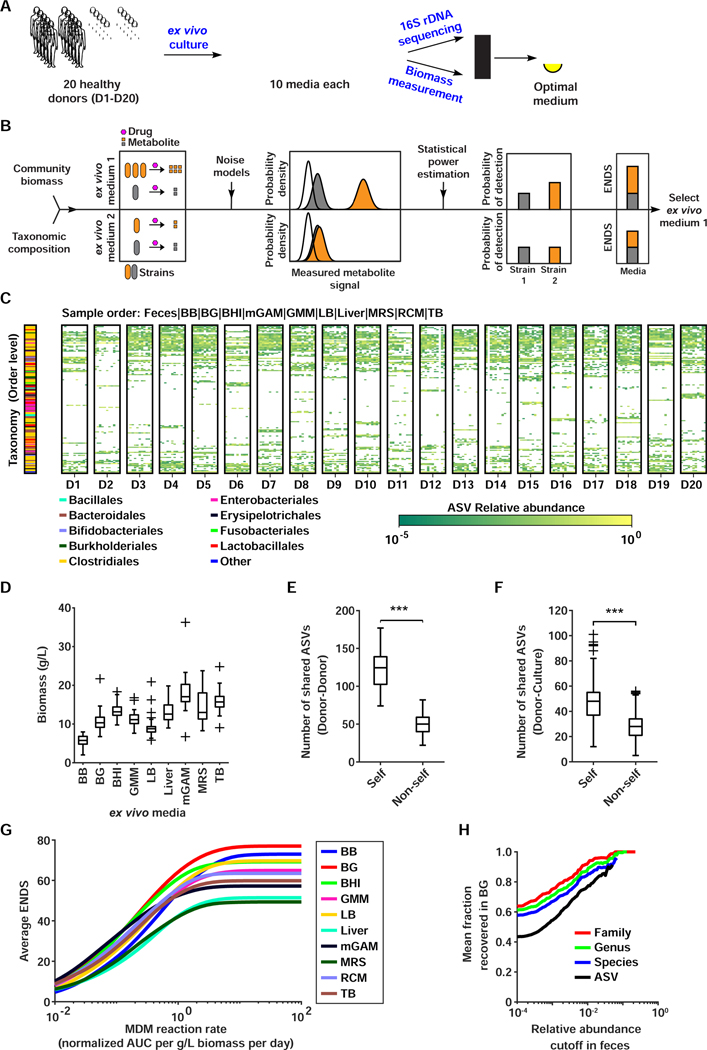
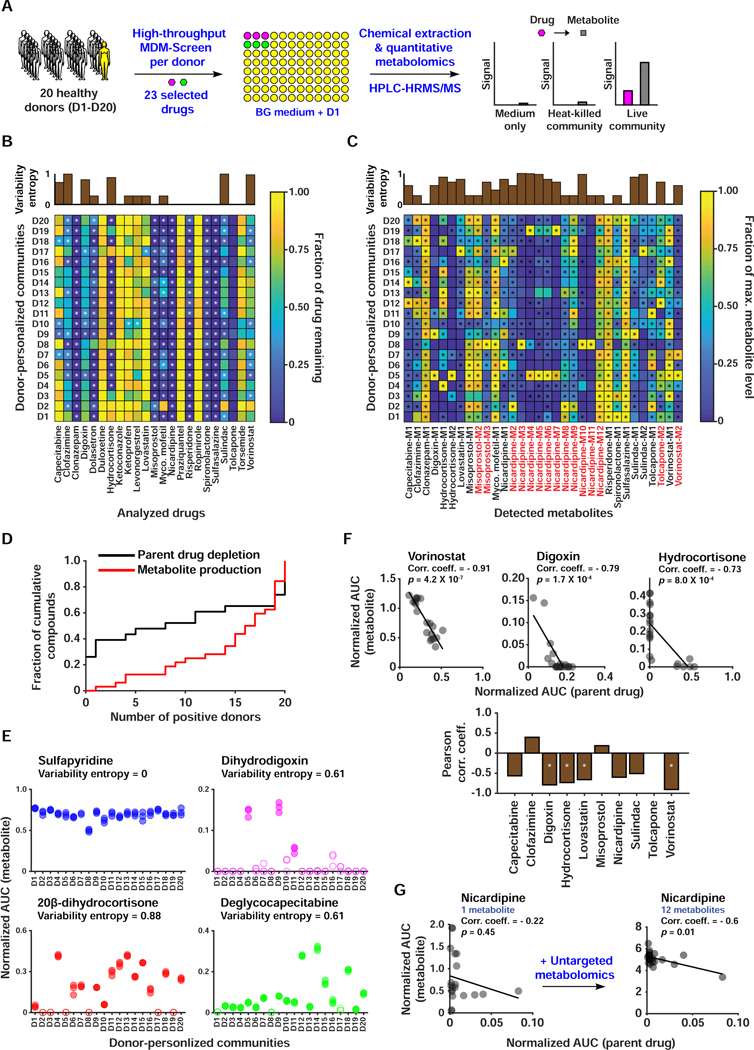
References
-
- Backhed F, Ley RE, Sonnenburg JL, Peterson DA, and Gordon JI (2005). Host-bacterial mutualism in the human intestine. Science 307, 1915–1920. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
