

RAS-targeted therapies: is the undruggable drugged?
- PMID: 32528145
- PMCID: PMC7809886
- DOI: 10.1038/s41573-020-0068-6
RAS-targeted therapies: is the undruggable drugged?
Erratum in
-
Author Correction: RAS-targeted therapies: is the undruggable drugged?Nat Rev Drug Discov. 2020 Dec;19(12):902. doi: 10.1038/s41573-020-0089-1. Nat Rev Drug Discov. 2020. PMID: 33082552
Abstract
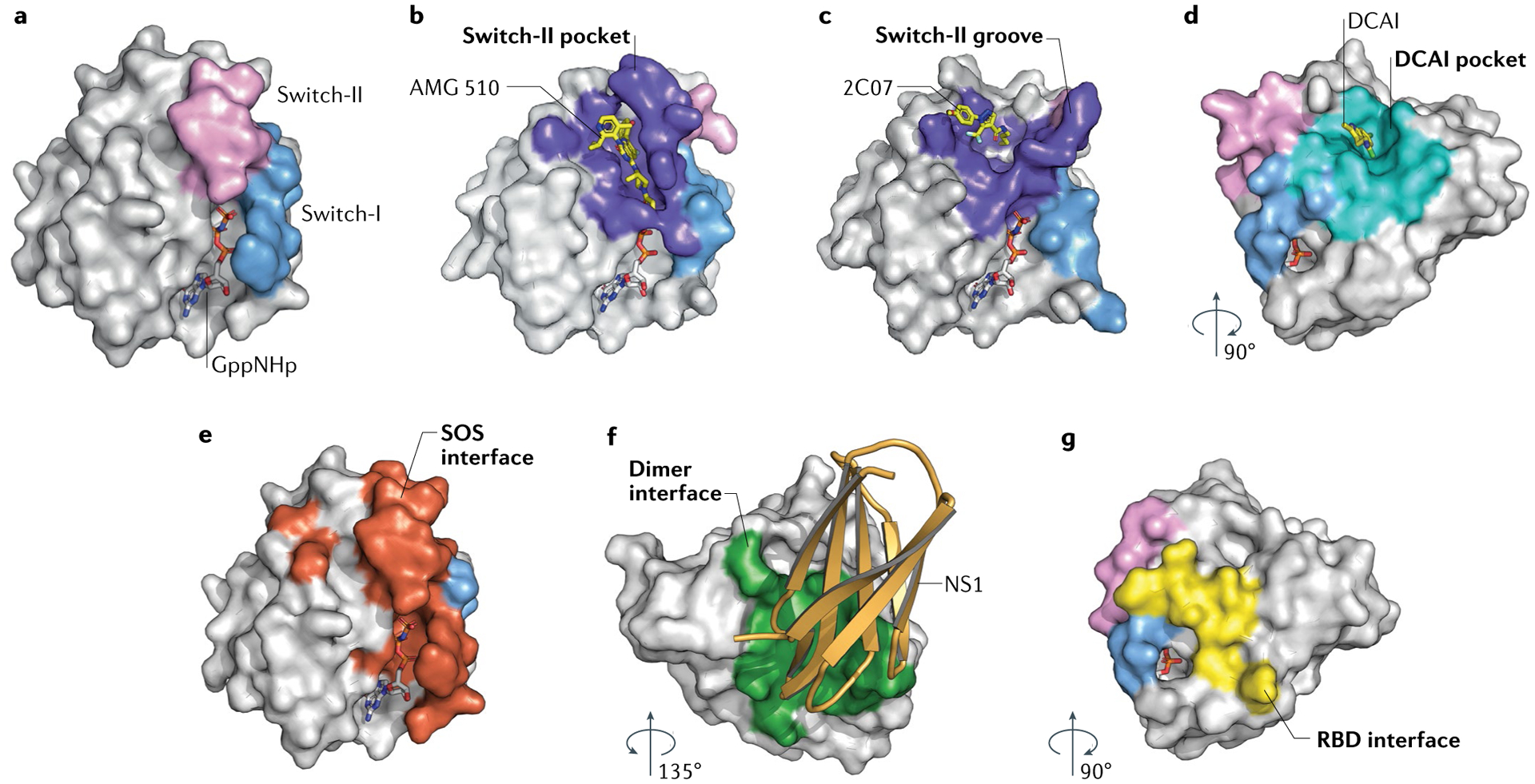
RAS (KRAS, NRAS and HRAS) is the most frequently mutated gene family in cancers, and, consequently, investigators have sought an effective RAS inhibitor for more than three decades. Even 10 years ago, RAS inhibitors were so elusive that RAS was termed 'undruggable'. Now, with the success of allele-specific covalent inhibitors against the most frequently mutated version of RAS in non-small-cell lung cancer, KRASG12C, we have the opportunity to evaluate the best therapeutic strategies to treat RAS-driven cancers. Mutation-specific biochemical properties, as well as the tissue of origin, are likely to affect the effectiveness of such treatments. Currently, direct inhibition of mutant RAS through allele-specific inhibitors provides the best therapeutic approach. Therapies that target RAS-activating pathways or RAS effector pathways could be combined with these direct RAS inhibitors, immune checkpoint inhibitors or T cell-targeting approaches to treat RAS-mutant tumours. Here we review recent advances in therapies that target mutant RAS proteins and discuss the future challenges of these therapies, including combination strategies.
Conflict of interest statement
Competing interests
F.McC. is a consultant for the following companies: Amgen, Pfizer Inc., and Quanta Therapeutics; is a consultant and co-founder with ownership interest including stock options of BridgeBio Pharma, Inc; and is Scientific Director of the NCI Ras Initiative at Frederick National Laboratory for Cancer Research/Leidos Biomedical Research Inc. S.M. is an employee of Genentech/Roche. A.R.M. and S.C.R. are also post-doctoral fellows employed by Genentech/Roche.
Figures
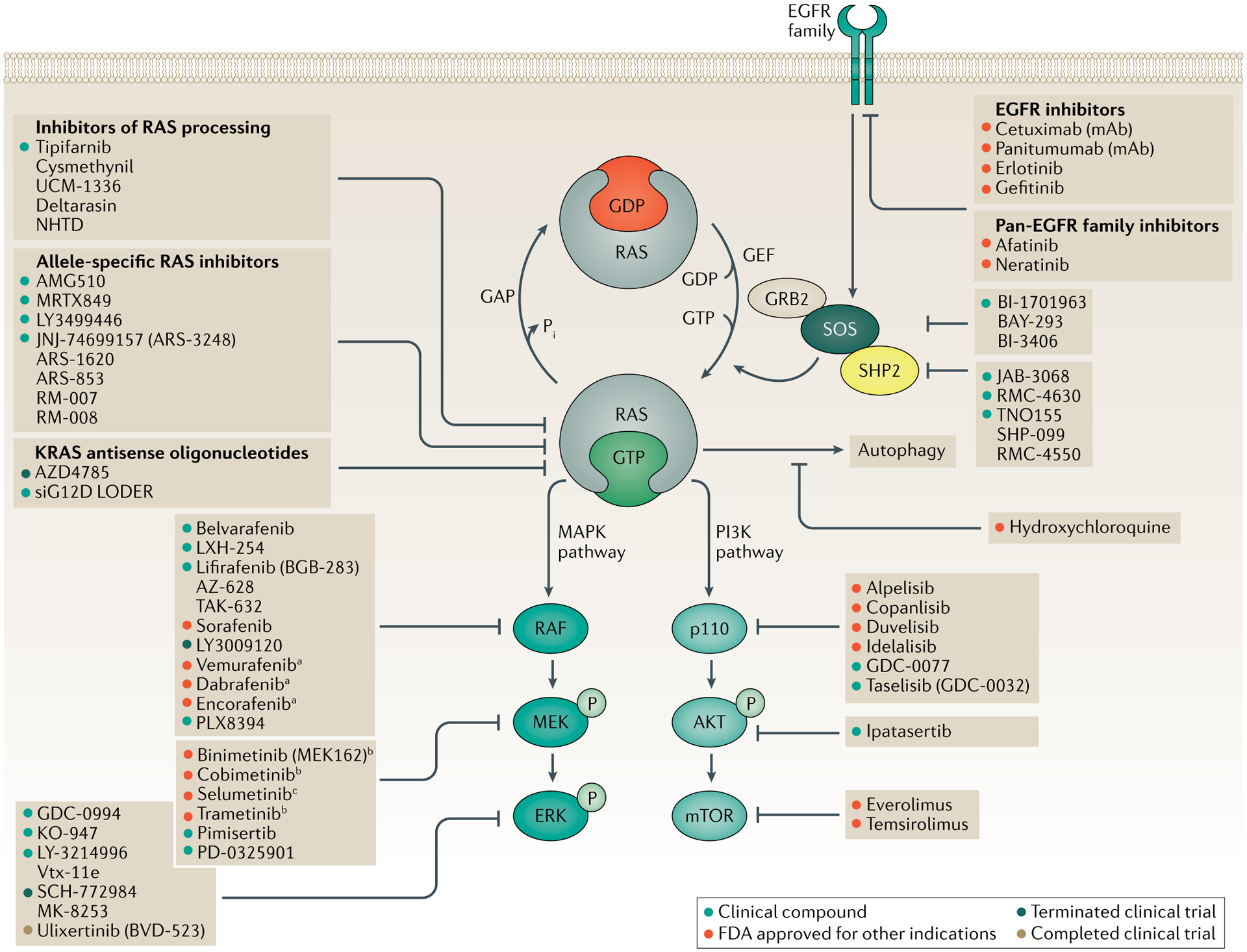
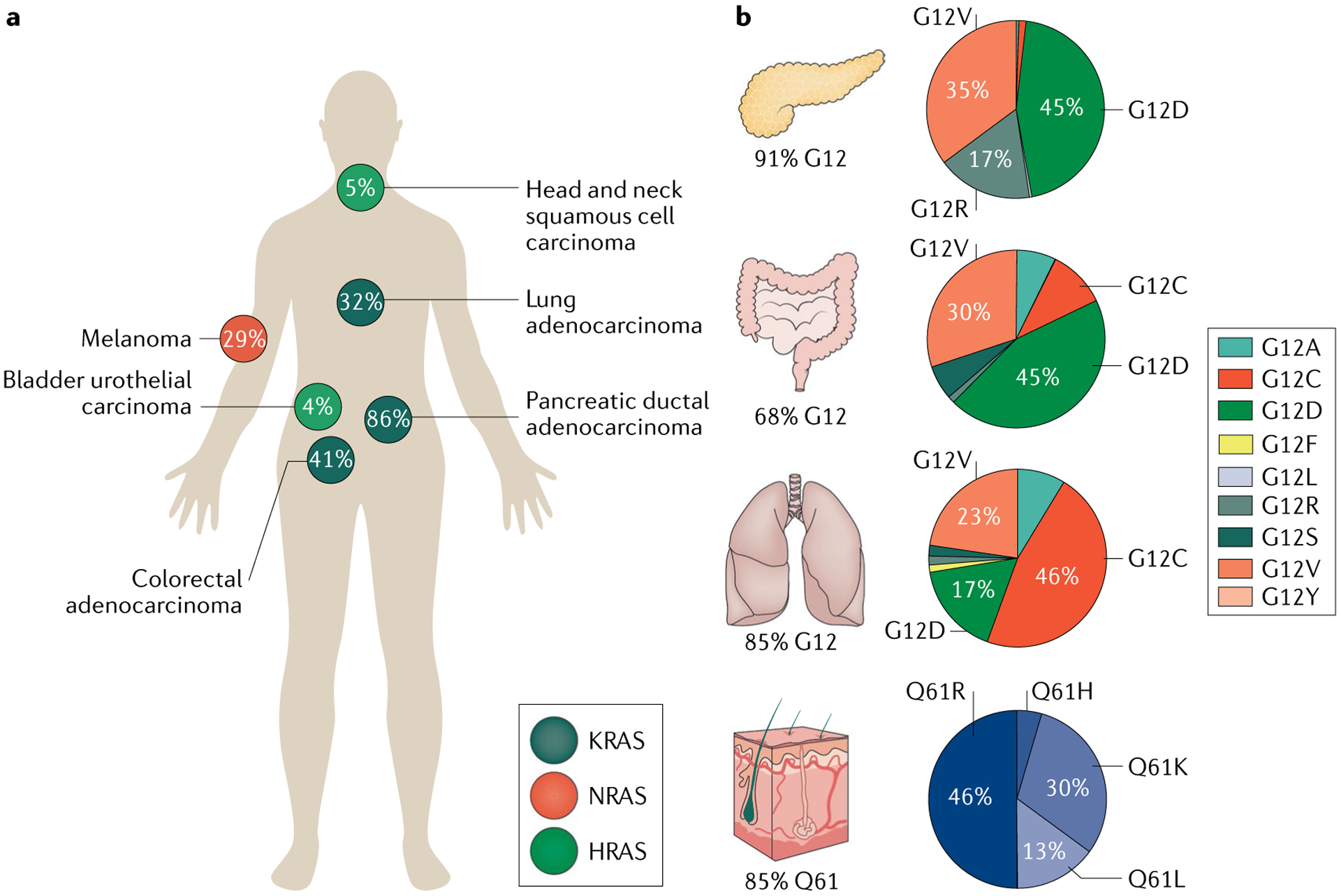
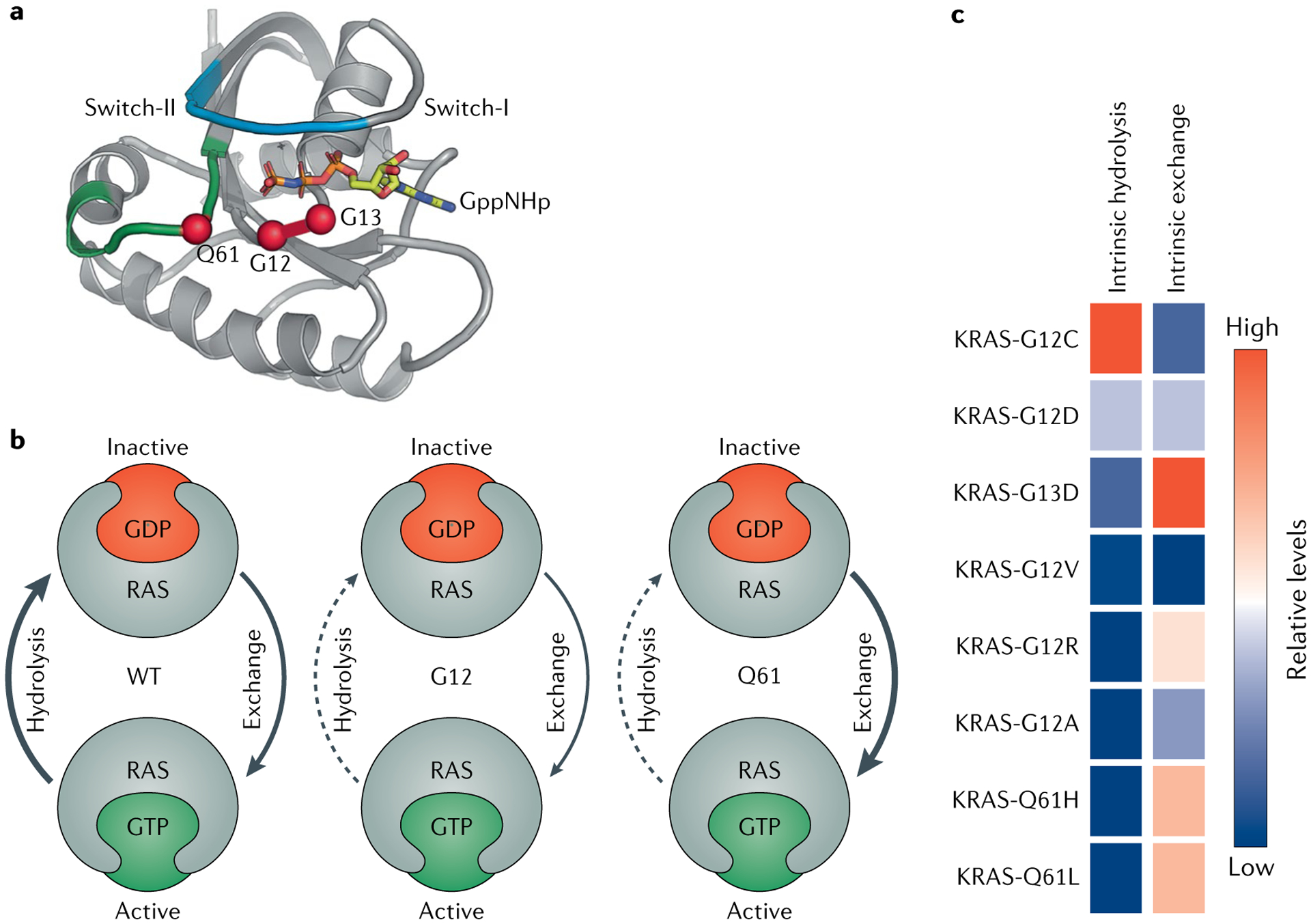
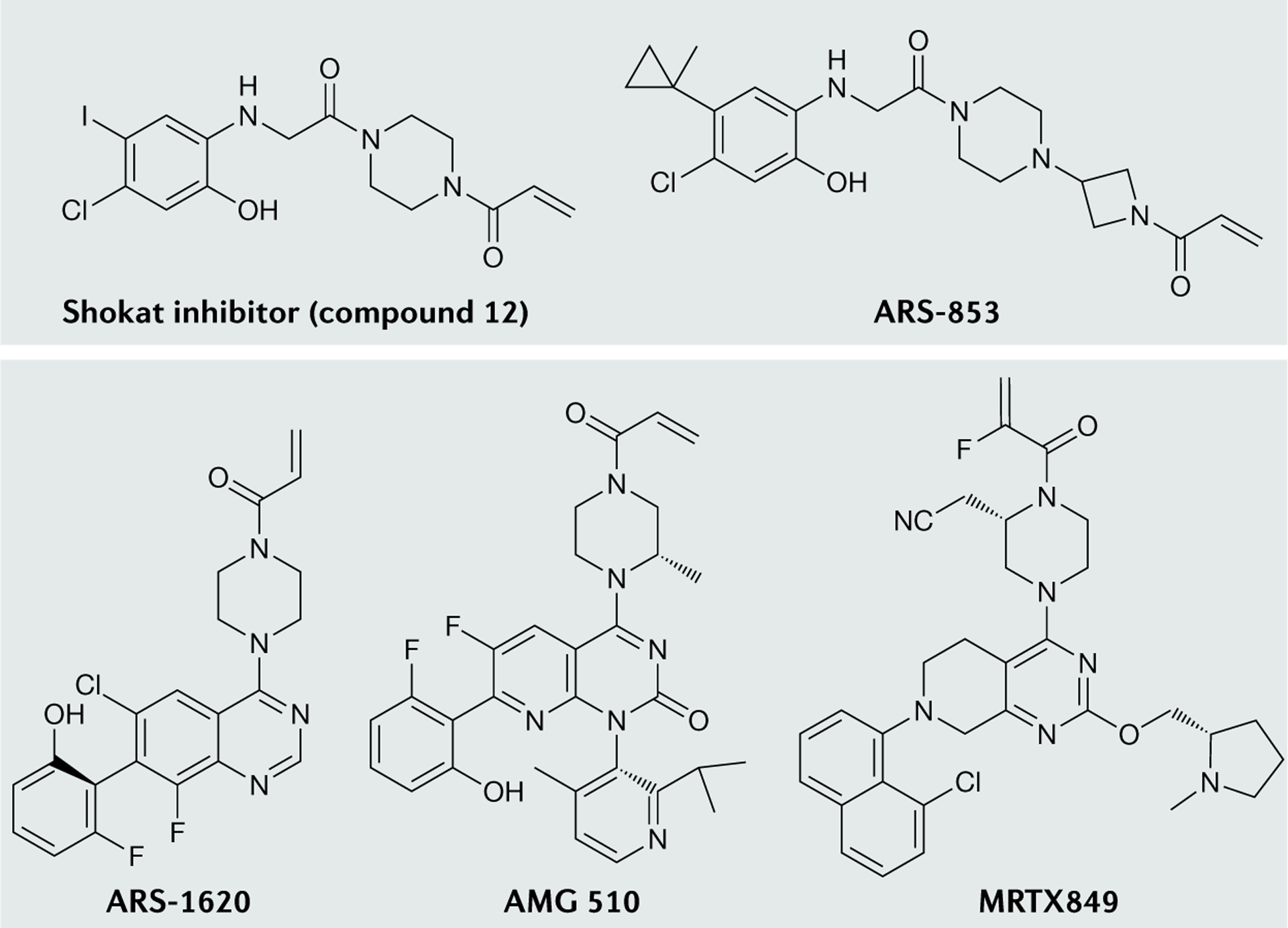
References
-
- Amgen. Amgen Announces New Clinical Data Evaluating Novel Investigational KRAS(G12C) Inhibitor in Larger Patient Group at WCLC 2019 www.amgen.com https://www.amgen.com/media/news-releases/2019/09/amgen-announces-new-cl... (2019).
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
