

Sequential targeted exome sequencing of 1001 patients affected by unexplained limb-girdle weakness
- PMID: 32528171
- PMCID: PMC7462745
- DOI: 10.1038/s41436-020-0840-3
Sequential targeted exome sequencing of 1001 patients affected by unexplained limb-girdle weakness
Abstract
Purpose: Several hundred genetic muscle diseases have been described, all of which are rare. Their clinical and genetic heterogeneity means that a genetic diagnosis is challenging. We established an international consortium, MYO-SEQ, to aid the work-ups of muscle disease patients and to better understand disease etiology.
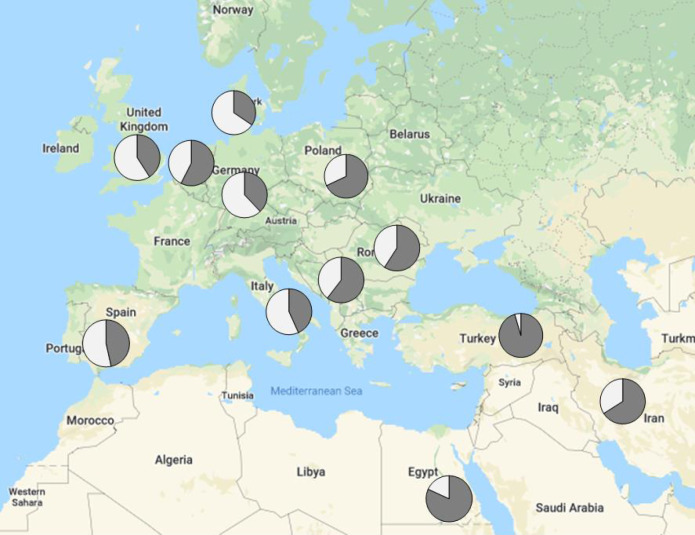
Methods: Exome sequencing was applied to 1001 undiagnosed patients recruited from more than 40 neuromuscular disease referral centers; standardized phenotypic information was collected for each patient. Exomes were examined for variants in 429 genes associated with muscle conditions.
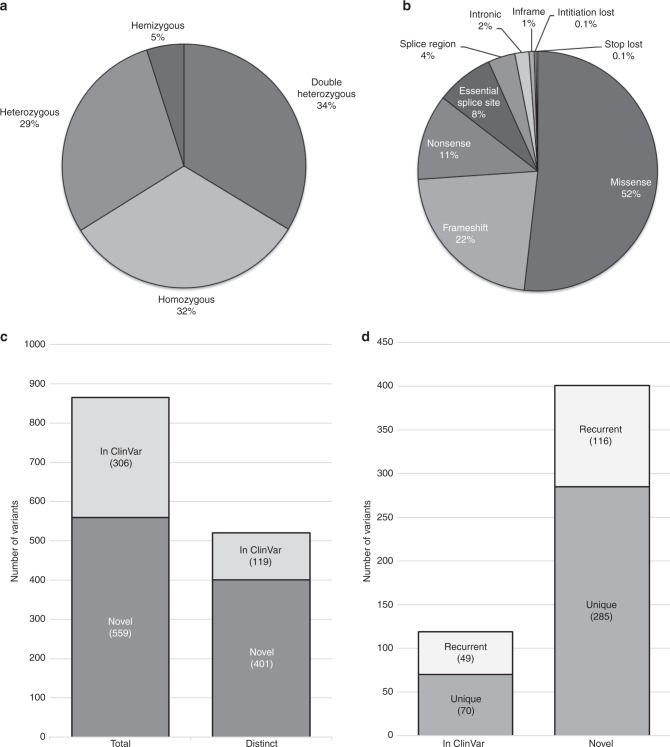
Results: We identified suspected pathogenic variants in 52% of patients across 87 genes. We detected 401 novel variants, 116 of which were recurrent. Variants in CAPN3, DYSF, ANO5, DMD, RYR1, TTN, COL6A2, and SGCA collectively accounted for over half of the solved cases; while variants in newer disease genes, such as BVES and POGLUT1, were also found. The remaining well-characterized unsolved patients (48%) need further investigation.
Conclusion: Using our unique infrastructure, we developed a pathway to expedite muscle disease diagnoses. Our data suggest that exome sequencing should be used for pathogenic variant detection in patients with suspected genetic muscle diseases, focusing first on the most common disease genes described here, and subsequently in rarer and newly characterized disease genes.
Keywords: genetic diagnosis; limb-girdle weakness; neuromuscular disease; next-generation sequencing; targeted exome analysis.
Conflict of interest statement
D.G.M. is a founder with equity in Goldfinch Bio, and has received research support from AbbVie, Astellas, Biogen, BioMarin, Eisai, Merck, Pfizer, and Sanofi Genzyme. V.S. is or has been a principal investigator for trials sponsored by Sanofi Genzyme, GSK, Prosensa/Biomarin, IonisPharmceuticals, and Sarepta. He has received speaker honoraria from Sanofi Genzyme. He is or has been on advisory boards for Acceleron Pharma, Audentes Therapeutics, Biomarin, Bristol-Myer Squibb, Italfarmaco S.p.A., Nicox, Pfizer, Sanofi Genzyme, Santhera, Sarepta Therapeutics, Summit Therapeutics, Tivorsan, and TrophyNOD. The other authors declare no conflicts of interest.
Figures

References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Miscellaneous
